

ФЕДЕРАЛЬНОЕ ГОСУДАРСТВЕННОЕ БЮДЖЕТНОЕ УЧРЕЖДЕНИЕ «НАУЧНЫЙ ЦЕНТР НЕВРОЛОГИИ» РОССИЙСКОЙ АКАДЕМИИ МЕДИЦИНСКИХ НАУК
На правах рукописи
РЫБАКОВА ЮЛИЯ СЕРГЕЕВНА
Антипролиферативное действие карнозина и его производных на опухолевые клетки нейрального происхождения
03.01.04 – биохимия
Диссертация на соискание ученой степени кандидата биологических наук
Научный руководитель:
доктор биологических наук
Федорова Татьяна Николаевна
МОСКВА 2013
СОДЕРЖАНИЕ
ГЛАВА 1. ОБЗОР ЛИТЕРАТУРЫ. 11
РАЗДЕЛ 1.1. Редокс-статус клетки определяется соотношением внутриклеточных про- и антиоксидантов. 11
1.1.1. Активные формы кислорода и их значение в жизни клетки. 11
1.1.2. Основные представители антиоксидантной системы клетки и их роль в поддержании редокс-статуса. 13
РАЗДЕЛ 1.2. Редокс-регуляция клеточной пролиферации. 17
1.2.1. Клеточный цикл и его регуляция. 18
1.2.2. Редокс-регуляция клеточного цикла. 26
РАЗДЕЛ 1.3. Современное состояние знаний о противоопухолевом эффекте короткоцепочечных пептидов. 36
1.3.1. Карнозин и его производные. Общая характеристика. 36
1.3.2. Антиоксидантные свойства карнозина и его производных. 39
1.3.3. Эффект карнозина и его производных на пролиферацию нормальных и опухолевых клеток. 42
1.3.4. Пинеалон. Общая характеристика и свойства. 45
ГЛАВА 2. МЕТОДЫ ИССЛЕДОВАНИЯ. 47
2.1. Культуры клеток. 47
2.2. Исследуемые соединения. 48
2.3. Исследование клеточной пролиферации. 48
2.3.1. Время удвоения популяции. 48
2.3.2. Эффективность посева и доля выживших клеток. 49
2.4. Проточная цитометрия. 49
2.4.1. Измерение уровня АФК. 50
2.4.2. Определение доли мертвых клеток. 51
2.4.3. Анализ клеточного цикла. 51
2.4.4. Двухпараметрический анализ клеточного цикла. 53
2.5. Иммуноблоттинг. 53
2.6. Определение активности MnСОД. 54
2.7. Определение активности каталазы. 54
2.8. Полимеразная цепная реакция (ПЦР) в режиме реального времени. 55
2.9. Статистическая обработка результатов. 55
ГЛАВА 3. РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ. 56
РАЗДЕЛ 3.1. Изучение характера действия карнозина на пролиферацию клеток. 56
3.1.1. Карнозин снижает количество клеток РС-12 в культуре. 56
3.1.2. Карнозин снижает уровень АФК в клетках культуры РС-12. 58
3.1.3. Карнозин и его производные индуцируют накопление клеток РС-12 в S и G2/М фазах клеточного цикла. 59
3.1.4. Изучение воздействия синтетического трипептида пинеалона на клеточный цикл. 63
РАЗДЕЛ 3.2. Изучение механизма регуляции клеточного цикла под действием карнозина. 66
3.2.1. Карнозин избирательно ингибирует пролиферацию клеток глиобластомы. 66
3.2.2. Карнозин снижает уровень АФК и индуцирует экспрессию MnСОД. 70
3.2.3. Карнозин индуцирует G2 блок и усиливает экспрессию циклина В1. 74
РАЗДЕЛ 3.3. Изучение функциональной нагрузки структурных элементов в молекуле карнозина при антипролиферативном эффекте. 78
РАЗДЕЛ 3.4. Возможности применения карнозина в радиотерапии. 82
ГЛАВА 4. ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ 83
ВЫВОДЫ 96
СПИСОК ЛИТЕРАТУРЫ 97
ВВЕДЕНИЕ
Карнозин представляет собой природный дипептид, состоящий из двух аминокислот – -аланина и L-гистидина. В организме человека карнозин представлен во многих тканях (мышцы, мозг, печень, ткани глаза, желудок, почки, легочная ткань) достигая наивысшей концентрации в скелетных мышцах (~ 20 мM) и в обонятельных луковицах головного мозга (~ 2.5 мM) [1, 2]. Карнозин синтезируется с помощью фермента карнозин-синтетазы, которая катализирует образование пептидной связи между -аланином и L-гистидином используя энергию АФТ и ионы Mg2+ с использованием энергии АТФ [3-7]. Разрушение карнозина на составляющие его аминокислоты осуществляется главным образом сывороточной карнозиназой (CN1) [8, 9].
Карнозин демонстрирует многообразие функций, приведем некоторые из них. Во время интенсивной мышечной работы карнозин выполняет функцию протонного буфера, связывающего избыток протонов, образующихся вместе с молочной кислотой, и препятствующего развитию ацидоза [10]. Карнозин также способен образовывать комплексы с металлами переменной валентности (Cu2+, Zn2+, Fe2+ и др.) [11-13]. Антиоксидантные свойства карнозина заключаются в его способности образовывать комплексы с двумя высоко реакционноспособными окислителями – гидроксильным радикалом и супероксидным анионом, снижая их реактивность и предотвращая дальнейшее распространение окислительного повреждения [14-17]. Кроме того, в нескольких независимых исследованиях было продемонстрировано эффективное подавление накопления продуктов перекисного окисления липидов (малоновый диальдегид, пероксильные радикалы, ненасыщенные альдегиды, 4-гидроксиноненаль) в присутствии карнозина, что снижало уровень окислительного повреждения макромолекул, способствуя сохранению их структуры и функций [15, 18, 19].
Антипролиферативный эффект карнозина на опухолевые клетки был впервые описан Nagai и Suda в 1986 году [20]. В их работе подкожные инъекции карнозина мышам линии ddY, с предварительно вживленными клетками саркомы, способствовали снижению интенсивности пролиферации опухолевых клеток и увеличению выживаемости животных. Позже в независимом исследовании Renner et al. на мышах с ксенографтами HER2/neu NIH3T3 фибробластов показали, что ежедневные внутрибрюшинные инъекции карнозина существенно подавляют пролиферацию злокачественных клеток и опухолевый рост [21]. Holliday и McFarland продемонстрировали избирательность действия карнозина на опухолевые клетки. Добавление карнозина к смеси клеток карциномы (HeLa) и нормальных человеческих фибробластов (MRC-5) приводило к выборочному уничтожению HeLa клеток и удлиняло продолжительность жизни фибробластов [22, 23]. Обработка карнозином нормальных эмбриональных стволовых клеток и злокачественных клеток эмбриональной терато-карциномы также приводило к избирательной гибели последних. Полученные данные указывали на то, что ни степень дифференцировки, ни скорость пролиферации клеток не являются определяющими факторами избирательного действия карнозина на опухолевые клетки. Несмотря на наличие большого количества экспериментальных данных об антипролиферативном действии карнозина на опухолевые клетки, механизм противоопухолевого эффекта остается не выясненным.
На сегодняшний день в литературе накоплено множество данных о том, что интенсивность пролиферации зависит от соотношения про- и антиоксидантов внутри клетки [24, 25]. Показано, что быстро пролиферирующие клетки характеризуются более высоким содержанием прооксидантов и пониженной активностью антиоксидантной системы по сравнению с клетками, делящимися медленно или находящимися в состоянии покоя [26, 27]. Было показано, что уровень прооксидантов в опухолевых клетках часто превышает прооксидантный уровень в нормальных клетках того же типа, что возможно является причиной их чрезмерной пролиферации [28, 29]. Добавление небольших концентраций прооксиданта пероксида водорода усиливало пролиферацию клеток [28], а повышение экспрессии антиоксидантного фермента MnСОД, наоборот, приводила к замедлению пролиферации [30]. Поскольку карнозин демонстрирует эффективные антиоксидантные свойства, мы предположили, что ингибирование пролиферации опухолевых клеток происходит в результате снижения уровня внутриклеточных прооксидантов под действием карнозина.
Работа выполнена в соответствии с планом НИР ФГБУ «НЦН» РАМН в рамках темы №146 «Исследование в условиях in vitro и in vivo проблем эффективности безопасности наноструктурных комплексов, обладающих нейропротекторным действием».
Цель исследования. Изучение механизма антипролиферативного действия карнозина и его производных на опухолевые клетки нейрального происхождения.
Задачи исследования:
1. Изучить характер воздействия карнозина на пролиферацию культур опухолевых клеток феохромоцитомы крысы (РС-12), карциномы горла и рта (FaDu, Cal27) и молочной железы (MB231) человека, глиобластомы человека (U-118-MG);
2. Выявить, сопровождаются ли индуцируемые карнозином изменения пролиферации опухолевых клеток колебаниями внутриклеточного уровня АФК и антиоксидантных ферментов;
3. Выяснить, являются ли индуцируемые карнозином изменения пролиферации опухолевых клеток результатом модификации прогрессии клеточного цикла;
4. Сравнить антипролиферативные свойства карнозина с действием его производных, а также синтетического трипептида пинеалона;
5. Оценить эффект совместного применения карнозина и ионизирующего излучения на гибель опухолевых клеток.
Научная новизна. Настоящая работа представляет собой оригинальное экспериментальное исследование, в котором было впервые показано избирательное подавление пролиферации клеток глиобластомы под действием карнозина. Установлено, что замедление пролиферации глиобластомы сопровождается накоплением клеток в G2 фазе клеточного цикла и активацией экспрессии циклина B1. Продемонстрировано, что параллельно с изменениями в прогрессии клеточного цикла происходит повышение активности MnСОД и понижение внутриклеточного уровня АФК. Выявлено, что метилированное производное карнозина – анзерин – подавляет пролиферацию глиобластомы эффективнее, чем карнозин. Показано, что предварительная инкубация клеток с карнозином снижает выживаемость клеток глиобластомы под действием ионизирующего излучения.
Теоретическая и практическая значимость работы. Полученные результаты существенно расширяют представления о природе антипролиферативного действия карнозина на опухолевые клетки нейрального происхождения, что важно для понимания молекулярных механизмов действия карнозина в целом. Полученные в работе данные об эффекте совместного действия карнозина и ионизирующего излучения на выживаемость клеток глиобластомы открывают перспективу для применения карнозина в комбинированной терапии опухолей головного мозга.
Основные положения, выносимые на защиту:
1. Карнозин ингибирует пролиферацию опухолевых клеток нейрального происхождения, при этом наиболее выраженный эффект проявляется на клетках глиобластомы человека U-118-MG;
2. Ингибирование пролиферации клеток глиобластомы под действием карнозина сопровождается снижением уровня АФК и увеличением активности MnСОД;
3. Изменения в антиоксидантной системе клеток глиобластомы сопровождаются накоплением клеток в G2 фазе клеточного цикла и усилением экспрессии циклина В1;
4. Метилированное производное карнозина, анзерин, ингибирует пролиферацию клеток глиобластомы эффективнее, чем карнозин;
5. Предварительная инкубация клеток с карнозином снижает выживаемость клеток глиобластомы под действием ионизирующего излучения.
Протокол диссертационного исследования «Антипролиферативное действие карнозина и его производных на опухолевые клетки нейрального происхождения» было одобрено локальным этическим комитетом ФГБУ «НЦН» РАМН. Протокол №12/13 от 11.12.2013.
Апробация работы. Диссертация апробирована и рекомендована к защите на совместном заседании научных сотрудников ФГБУ «Научный центр неврологии» РАМН 18 октября 2013 года.
Материалы диссертационной работы были представлены на V Stromboli Conference on Cancer and Ageing: “The Primeval Life-Generating Molecules. Therapeutic and Aging-Reversing Properties” (Стромболи, Италия, 2010) и The 4th Quadrennial Meeting of the World Federation of Neuro-Oncology held in conjunction with the 2013 SNO Scientific Meeting and Education Day (Сан-Франциско, США, 2013)
Публикации. По материалам диссертации опубликовано 4 научные работы, из них 1 публикация в изданиях, рекомендуемых ВАК Министерства образования и науки РФ и 2 работы в зарубежных рецензируемых журналах.
Личный вклад автора. Автором лично выполнено культивирование клеточных линий и исследования клеточной пролиферации, проведено измерение уровня АФК, доли мертвых клеток, а также анализ клеточного цикла, определена активность и экспрессия антиоксидантных ферментов. Выполнена последующая аналитическая обработка и обобщение полученных результатов, сформулированы выводы и подготовлены публикации.
Структура и объем диссертации. Диссертация состоит из введения, обзора литературы, описания материалов и методов исследования, изложения результатов, их обсуждения, выводов и списка литературы. Работа содержит 120 стр. машинописного текста, 1 таблицу и 29 рисунков. Список литературы включает 271 источник (18 отечественных и 253 зарубежных).
ГЛАВА 1. ОБЗОР ЛИТЕРАТУРЫ
РАЗДЕЛ 1.1. Редокс-статус клетки определяется соотношением внутриклеточных про- и антиоксидантов
1.1.1. Активные формы кислорода и их значение в жизни клетки
Наиболее распространенные и реакционноспособные прооксиданты в клетке – активные формы кислорода (АФК). АФК представляют собой продукты частичного восстановления кислорода, содержащие один или несколько неспаренных электронов, и относятся к классу свободных радикалов [31].
Главным источником АФК в клетке является дыхательная цепь митохондрий [32], где теряется порядка 2% супероксида. Помимо этого АФК образуются при работе некоторых ферментов (НАДФН-окcидаза [33], ксантиноксидаза [34], монооксигеназа, NO-синтаза [35] и др), метаболизме ксенобиотиков, активации дыхательного взрыва в лейкоцитах, под воздействием цитокинов, ионизирующего и УФ-излучения [36]. Образование АФК происходит в несколько стадий. Одноэлектронное восстановление кислорода приводит к формированию супероксидного аниона (или супероксида, О2•-), который сам по себе не является окислителем, а наоборот, по свойствам напоминает слабое основание. Поэтому при физиологическом pH супероксид присутствует частично в протонированной форме (НО2•, pKa = 4.8), которая является достаточно сильным окислителем [37]. НО2• представляет собой, по сути дела, пероксильный радикал (ROO•) без радикала (R), и поэтому подобно ROO•, легко инициирует перекисное окисление липидов (ПОЛ), приводя к образованию новых форм АФК: продуктов ПОЛ, пероксильных (ROO•) и алкоксильных (RO•) радикалов [38]. Супероксид достаточно быстро дисмутирует с образованием пероксида водорода (Н2О2): О2-• + 2HО2• 2O2 + H2O2, (k=9,7x107 M-1•s-1) [39]. Взаимодействие между супероксидом и пероксидом водорода (реакция Фентона) приводит к образованию гидроксильного радикала (HO•): 2О2-• + H2О2 + 2H+ 2HO• + 2HO- + O2. Сама по себе реакция идет очень медленно (k<0.1 M-1•s-1), однако значительно ускоряется в присутствии ионов переходных металлов (Fe2+, Cu2+ и др.) [40]. HO• является самым сильным окислителем в водных растворах, реагируя с огромной скоростью с любыми находящимися по близости молекулами (k 109 – 1010 M-1•s-1 для соединений содержащих C, H, O, S, N) [37]. Описанные выше превращения можно обобщить в виде четырех простых реакций:
О2 + 1е- + (H+) О2•- (НО2•)
НО2•- + 1е- + H+ Н2О2
Н2О2 + 1е- + Н+ Н2О + НО•
НО• + 1е- + H+ Н2О
В результате воздействия ионизирующего или УФ-излучения, некоторых химических соединений (ксенобиотики и др.), а также при старении и опухолевой трансформации происходит повышение внутриклеточного уровня АФК, что приводит к окислительному повреждению макромолекул (ДНК, белки, липиды), нарушению их структуры, функции и в итоге к гибели клетки. В то же время, на сегодняшний день накоплено множество данных о том, что в небольших количествах АФК играют важную роль в жизни клетки [41-44]. В независимых исследованиях было продемонстрировано участие АФК в регуляции пролиферативных процессов. Добавление O2•- к среде культивирования нормальных фибробластов или лимфоцитов усиливало пролиферацию клеток [45, 46]. Laurent et al. показали, что небольшие концентрации Н2О2 (0,02 – 0,13 мкМ) индуцируют пролиферацию нормальных мышиных фибробластов NIH 3T3, а также опухолевых клеток линий СТ26 (карцинома прямой кишки мыши) и Нера 1-6 (гепатома мыши) [28]. Сравнительный анализ образования АФК в нормальных и опухолевых клетках выявил, что уровень АФК в опухолевых клетках значительно выше по сравнению с нормальными клетками того же вида [29]. Образование АФК в нормальных клетках происходит в главным образом за счет работы НАДФН-оксидаз, в то время как основным источником АФК в опухолевых клетках являются митохондрии [28].
1.1.2. Основные представители антиоксидантной системы клетки и их роль в поддержании редокс-статуса
Концентрация внутриклеточных прооксидантов регулируется антиоксидантной системой клетки, которая включает как низкомолекулярные вещества (витамин Е, С, -каротин, глутатион, НАДФН, пируват, тиоредоксин и др.), так и ферменты (супероксиддисмутаза, глутатионпероксидаза, каталаза, тиоредоксин пероксидаза и др.). Таким образом, в норме в клетке постоянно поддерживается равновесие между скоростью образования прооксидантов и их устранением с помощью системы антиоксидантов. Это равновесие называется окислительно-восстановительным балансом или редокс-статусом [47].
Самым распространенным низкомолекулярным антиоксидантом клетки является цистеин-содержащий трипептид - глутатион (Gly-Cys-Glu, GSH), его концентрация в цитоплазме может достигать 11 мМ [48]. Благодаря наличию чувствительной к окислению -SH группы цистеина GSH способен улавливать малейшие изменения редокс-статуса и реагировать на них. Соотношение восстановленной формы глутатиона (GSH) и окисленной (GSSH) часто используется для оценки состояния внутриклеточного окислительно-восстановительного баланса [47, 49].
Одним из основных антиоксидантных ферментов клетки является супероксиддисмутаза (СОД), катализирующая реакцию дисмутации супероксида: О2-• + О2-• O2 + H2O2. Несмотря на то, что дисмутация О2-• происходит быстро и сама по себе, СОД делает реакцию практически молниеносной (k=2х109 M-1•s-1) [50]. Cуществует три изоформы СОД: митохондриальная (MnСОД) [50], цитоплазматическая (CuZnСОД) [51] и внеклеточная, на поверхности клеточных мембран, (ЕС(CuZn)СОД) [52]. Поскольку митохондрии являются главным источником АФК в клетке, митохондриальная MnСОД представляется первым барьером защищающим клетку от окислительного повреждения. Несколькими независимыми лабораториями было показано, что полное ингибирование MnСОД (Sod2-/-) у новорожденных мышей приводит к гибели животных в течение 1-18 дней после рождения и сопровождается дилатационной кардиомиопатией, усилением накопления липидов в печени и метаболическим ацитодозом [53, 54]. Ингибирование экспрессии других изоформ СОД, каталазы или глутатионпероксидазы было совместимо с жизнью. Частичное ингибирование активности MnСОД (Sod2+/-) приводило к нарушению функционирования митохондрий и усилению их окислительного повреждения, а также увеличению риска развития опухолей [55-57]. Окислительное повреждение митохондрий и нарушение их функции, видимо, и являются причиной усиления образования митохондриальных АФК в опухолевых клетках. Пероксид водорода, образующийся в результате дисмутации О2-•, утилизируется в пероксисомах с помощью каталазы (H2O2 + H2O2 H2O + O2) [58, 59] или в цитоплазме с помощью глутатионпероксидазы (GPx) (H2O2 + 2GSH 2H2O + GSSG). Причем, GPx катализирует реакцию не только с H2O2, но и с ROOH приводя к образованию спиртов (ROH) [60, 61].
Благодаря способности модулировать внутриклеточный уровень АФК антиоксиданты способны влиять на пролиферацию клеток. Как было отмечено выше (см. 1.1.1), в небольших количествах АФК индуцируют пролиферацию клеток. Снижение уровня Н2О2 за счет усиления экспрессии каталазы в эндотелиальных клетках и HER-2/Neu-трансформированных Rat-1 фибробластах приводило к ингибированию пролиферации этих клеток [62, 63]. Понижение уровня АФК под действием синтетического антиоксиданта N-ацетил-цистеина (NAC) в клетках слизистой оболочки полости рта тажке приводило к замедлению пролиферативных процессов [64, 65].
Исследование пролиферации эмбриональных мышиных фибробластов с различным уровнем экспрессии MnСОД выявило, что пролиферация клеток содержащих MnСОД (+/+) замедляется по мере увеличения плотности популяции (контактное торможение). Частичное подавление экспрессии MnСОД приводило к тому, что MnСОД (+/-) клетки были нечувствительны к контактному торможению и продолжали пролиферировать. Рост MnСОД (-/-) фибробластов был существенно замедлен. Усиление экспрессии MnСОД в MnСОД (+/-) клетках приводило к замедлению пролиферации. Ингибирование MnСОД в клетках экспрессирующих MnСОД (+/+), наоборот, приводило к усилению пролиферации. Интересно отметить, что обработка MnСОД (+/-) клеток с помощью Tiron, ловушки для O2-•, приводила к понижению уровня O2-• и замедлению пролиферации, указывая на важную роль O2-• в поддержании пролиферации. Замедление пролиферации также сопровождалось повышением концентрации Н2О2, продукта работы MnСОД [66]. Исследование посттрансляционных модификаций в молекуле MnСОД выявило различия в сайтах метилирования в пролиферирующих и покоящихся клетках. Уровень метилирования MnСОД в активно пролиферирующих клетках был ниже, чем в клетках, пролиферация которых была замедленна [27].
Опухолевые клетки часто характеризуются смещением редокс в более окисленное состояние, что происходит в результате усиления образования АФК и ослабления работы антиоксидантных ферментов [67-70]. В отличие от нормальных клеток, в которых уровень GSH снижался по мере увеличения количества клеток (т.н. контактное торможение), уровень GSH в опухолевых клеток был постоянно высоким [71]. Вероятно, поэтому опухолевые клетки были невосприимчивы к действию NAC, предшественнику GSH. Обработка клеток карциномы молочной железы с помощью NAC не приводила к восстановлению редокс и формированию G1 блока [72]. Снижение внутриклеточного уровня GSH за счет ингибирования -глутамилцистеинсинтетазы с помощью DL-бутионин-S,R-сульфоксимина (BSO), приводило к смещению внутриклеточного редокс-статуса в более окисленное состояние и аресту клеточной пролиферации [73].
Активность антиоксидантных ферментов в опухолевых клетках часто понижена по сравнению с нормальными клетками того же типа что, вероятно, является одной из причин активной пролиферации опухолевых клеток. Например, активность CuZnСОД и MnСОД в нормальных клетках печени мыши составляла, соответственно, 122 и 35 ед/мг, в то время как, в клетках гепатомы активность CuZnСОД падала до 40 ед/мг, а активность MnСОД вообще не определялась [74]. Повышение экспрессии MnСОД в опухолевых клетках приводило к замедлению опухолевого роста [74-76]. Сходные данные были получены для каталазы и некоторых изоформ GPx. Снижение активности ферментов приводило к повышению уровня АФК, усилению пролиферативных процессов, а также способствовало развитию опухолевой трансформации [77, 78]
Резюмируя вышесказанное, важно отметить следующее: в клетке постоянно поддерживается баланс между про- и антиоксидантами. Наиболее распространенными прооксидантами являются АФК, к которым относятся: О2•-, Н2О2, НО•, ROO• и др. Основные регуляторы уровня внутриклеточных прооксидантов – антиоксиданты MnСОД, каталаза и GSH. Повышение уровня АФК приводит к гибели клетки, однако в небольших количествах АФК способны индуцировать пролиферативные процессы. Быстро делящиеся опухолевые клетки часто характеризуются повышенным уровнем АФК и пониженной активностью антиоксидантной системы, что возможно является одной из причин их активной пролиферации. Повышение антиоксидантной активности и понижение уровня АФК приводят к замедлению пролиферативных процессов, это открывает возможность контролировать пролиферацию опухолевых с помощью антиоксидантов.
РАЗДЕЛ 1.2. Редокс-регуляция клеточной пролиферации
Для того чтобы понять, механизм редокс-регуляции пролиферации, необходимо вспомнить - что такое пролиферация. Пролиферация - это процесс увеличения количества клеток, определяющийся соотношением скоростей деления клеток и их гибели. Гибель клеток может происходить как в результате повреждения целостности клетки (некроз), так и путем активации внутриклеточных программ (апоптоз). Деление клетки строго регулируется как внутренними программами, так и внешними факторами (факторы роста, цитокины и др.), воздействующими на внутренние программы. Каждому делению предшествует комплекс последовательных внутриклеточных изменений, т.н. клеточный цикл, основная цель которого – удвоение содержимого клетки и его равномерное распределение между двумя дочерними клетками при делении.
1.2.1. Клеточный цикл и его регуляция
Клеточный цикл представляет собой высокоорганизованную и строго регулируемую последовательность переходов от G1 фазы к S, к G2 и митозу (М). Переход от одной фазы к другой регулируется последовательной экспрессией белков циклинов и активацией циклин-зависимых киназ (cyclin dependent kinase, CDK). CDK представляют собой серин/треоининовые протеинкиназы, которые активируются при соединении с циклинами и запускают события клеточного цикла, путем фосфорилирования соответствующих остатков в белках-мишенях [79]. На Рис. 1.1. представлены основные циклин/CDK комплексы позвоночных, специфичные для той или иной фазы клеточного цикла. Уровень циклинов меняется по мере прогрессии клеточного цикла, приводя к колебаниям активности CDK (См. Рис. 1.1). Помимо циклинов активность CDK комплексов регулируется с помощью CDK-активирующей киназы (CDK-activating kinase, CAK), ингибиторов CDK (cyclin-dependent kinase inhibitor protein, CKI), а также протеолитическими процессами (убиквитинирование) [80, 81].
Для полной активации комплекса циклин/CDK необходимо фосфорилирование CDK по Thr161 (CDK1), Thr160 (CDK2) или Thr172/177 (CDK4/6), которое осуществляется в ядре с помощью CAK [83-85]. САК состоит из трех субъединиц: CDK7, циклин H и белка Mat1, который активирует комплекс циклин H/CDK7. Связывание с циклином приводит к конформационным изменениям в CDK, которые делают возможным фосфорилирование CDK с помощью CAK [86]. Активность CAK-киназы постоянна на протяжении всего клеточного цикла [87].
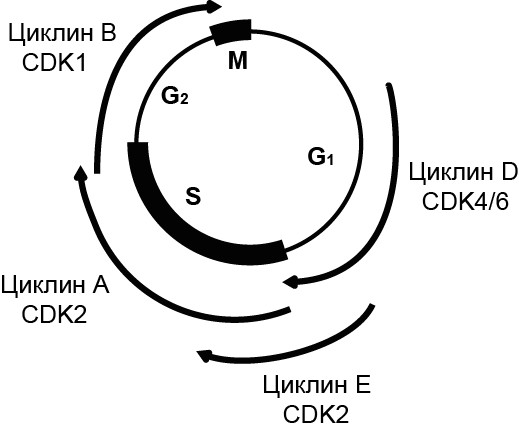
Рисунок 1.1. Основные фазо-специфические комплексы циклинов с циклин-зависимыми киназами у позвоночных. Из [82], с изменениями. См. пояснения в тексте.
На сегодняшний день насчитывается два семейства CKI: INK4 и Cip/Kip. Семейство INK4 включает белки р15, р16, р18 и р19, которые специфически связываются с CDK4/6, препятствуя образованию комплекса с циклином D [88, 89]. Увеличение уровня белка р16 часто наблюдается в стареющих клетках и сопровождается снижением пролиферативных способностей [90]. Белки р21, р27 и р57, относящиеся к Cip/Kip семейству, связываются с CDK2 и контролируют прогрессию через S фазу [91]. Ингибирование CKI часто приводит к нарушению контроля пролиферации и опухолевой трансформации [92-94].
Важную роль в регуляции протеолитических процессов во время клеточного цикла играет Комплекс стимулирующий анафазу (Anaphase promoting complex, АРС), который представляет собой убиквитин-лигазу, фермент катализирующий реакцию присоединения множественных копий белка убиквитина к боковым цепям лизина в молекуле белка-мишени [95, 96]. Полиубиквитинированние часто является сигналом к деградации белка, которое происходит в 26S протеасоме [97]. АРС присутствует в клетке на протяжении всего цикла, однако активность комплекса варьирует. АРС активируется в середине митоза, а ингибирование АРС происходит в конце G1 фазы [98, 99].
Некоторые химические соединения, ионизирующее и УФ излучение, ингибиторы репликации ДНК, а также ошибки при репликации или репарации ДНК приводят к нарушению структуры ДНК и остановке клеточного цикла. Главную роль в этом процессе играют 2 серин/треониновые протеин киназы – АТМ (ataxia telangiectasia mutated) и ATR (ATM-Rad3-related protein). АТМ активируется в ответ на появление двуцепочечных разрывов ДНК, образующиеся под воздействием ионизирующего облучения. ATR активирует под действием УФ-облучения или при нарушениях репликации ДНК. ATM и ATR прикрепляются к сайту повреждения ДНК и фосфорилируют различные белки, в том числе Chk1 и Chk2, которые приводят к остановке клеточного цикла в G1 фазе, препятствуя удвоению ДНК, или в G2 фазе, блокируя вход в митоз [100-103].
Инициация пролиферации. Сигнальные молекулы, стимулирующие деление клетки, называются митогенами. К наиболее распространенным митогенам относятся факторы роста (ФР; эпидермальный ФР (EGF), ФР фибробластов (FGF), ФР тромбоцитов (PDGF), инсулиноподобный фактор роста (IGF) и др.). Факторы роста связываются со специфическими рецепторами на поверхности клетки, приводя к их активации. Активированные рецепторы индуцируют внутриклеточные сигнальные каскады митоген-активируемых протеинкиназ (mitogen-activated protein kinases, МАРК). Существует 4 вида МАРК: ERK1/2, JNK, p38 и ERK5. Каскад ERK1/2 активируется в ответ на пролиферативные стимулы, например взаимодействие ФР с рецептором на поверхности мембраны. Каскады JNK, p38 и ERK5 активируются в основном при стрессе. ERK1/2 представляют собой консервативные серин/треониновые протеинкиназы, способные фосфорилировать различные белки-мишени в цитопламе, мембранах и ядре, и, таким образом, регулировать большое разнообразие внутриклеточных процессов [104, 105]. Показано, что активация ERK1/2 необходима для пролиферации клеток. Активированные ERK1/2 киназы принимают участие в синтезе пиримидинов, ремоделировании хроматина, синтезе новых рибосом, активации факторов трансляции и транскрипции, а также в регуляции G1/S и G2/M переходов между фазами клеточного цикла. Ингибирование ERK1/2 приводило к остановке пролиферации и накоплению клеток в одной из фаз клеточного цикла. Финальным шагом ERK1/2 киназ является активация факторов транскрипции (Myc, Elk-1, Sap-1, TIF-IA, и др.), которые индуцируют экспрессию генов, запускающих события клеточного цикла [106-109].
G1 фаза. В G1 фазе происходит активный рост новообразованной дочерней клетки, проверяется целостность и равномерность распределения хромосом после митоза. Первым под действием митотического сигнала активируется экспрессия циклинов D (в частности D1), которые образуют комплексы с CDK 4 или CDK 6 [110]. Главная роль комплекса циклин D1/CDK4/6 заключается в фосфорилировании белка ретинобластомы (retinoblastoma tumor suppressor protein, рRb) и активация фактора транскрипции E2F. В отсутствии митотической стимуляции E2F связан с pRb и поэтому не активен. Фосфорилирование pRb приводит к конформационным изменениям в молекуле pRb и освобождению E2F [111]. E2F связывается со специфической последовательностью ДНК и активирует экспрессию большого количества разнообразных генов необходимых для входа в S фазу, среди них циклины Е и А, ДНК полимераза, белок-ингибитор раннего митоза (early mitotic inhibitor, Emi1) и др. [112]. В конце G1 фазы циклин D1 разрушается, что приводит к ингибированию связанных с ним CDK. Разрушение циклина D1 происходит с помощью киназы гликогенсинтетазы (GSK-3), которая фосфорилирует циклин D1 по Thr286, или с помощью Mirk/dyrk киназы, которая фосфорилирует циклин D1 по Thr288. В обоих случаях фосфорилирование влечет за собой деградацию циклина в протеасомах [113, 114]. Ингибирование экспрессии циклина D1 препятствует переходу клеток в S фазу. В то время как эктопическая экспрессия циклина D1 ускоряет прогрессию через G1 фазу [115].
Интересно отметить, что для перехода из G1 фазы в S, также необходима активация ERK1/2 с последующей транслокацией протеинкиназы в ядро. Показано, что ERK1/2 индуцирует экспрессию циклина D1, предположительно, через активацию экспрессии генов семейства Fos и Myc. Экспрессия Fos запускается с помощью ERK-активируемого фактора транскрипции Elk. Фосфорилирование Myc по Ser62 с помощью ERK1/2 увеличивает стабильность Myc, который способен напрямую индуцировать экспрессию циклина D1 [106].
В конце G1 фазы находится т.н. точка «Старт» (Start или Restriction point) - контрольная точка, в которой оценивается готовность клетки и благоприятность внешних условий (наличие питательных веществ, факторов роста и др.) к переходу в S фазу и удвоению ДНК. Прошедшая через «Старт» клетка – запрограммирована на репликацию и больше не зависима от внешних стимулов.
К концу G1 фазы возрастает количество циклина Е, который образует комплекс с CDK2. Фосфатаза Cdc25A дефосфорилирует CDK2 по p-Tyr15 и p-Thr14, активируя комплекс циклин Е/CDK2, необходимый для продвижения клетки из G1 в S [116]. Как только клетки входят в S фазу циклин Е деградирует и CDK2 соединяется с циклином А [81, 110]. Циклин А не накапливается в клетке вплоть до поздней G1 фазы из-за убиквитинирования под действием АРС. Циклины D и Е нечувствительны к действию АРС. Перед входом в S фазу Emi1, экспрессия которого запускается с помощью E2F, ингибирует АРС, способствуя стабилизации циклина А и активации СDK2. [117].
S фаза. Основным событием S фазы является удвоение ДНК (репликация) и хроматина. Репликация может начинаться только в определенных местах – точках начала репликации (origins of replication, OR), которые разбросаны по всей ДНК. Подготовка к репликации начинает еще в конце митоза – начале G1 фазы, когда в точках начала репликации собираются т.н. пререпликационные комплексы (pre-RC), состоящие из большого количества белков необходимых для инициации репликации. Эта стадия часто называется лицензированием (licensing) точек начала репликации, т.к. синтез ДНК может происходить только в OR содержащих pre-RC. В S фазе активный комплекс циклин А/CDK2 запускает сборку преинициирующих комплексов вокруг компонентов pre-RC. Преинициирующие комплексы раскручивают спираль ДНК подготавливая место посадки ДНК, полимеразы и других ферментов репликации. Сразу после репликации OR комплекс pre-RC разбирается под действием циклин А/CDK2. Сборка новых pre-RC в S фазе невозможна из-за присутствия активных СDK, что препятствует множественной репликации [118, 119]. Репликация хромосом состоит из репликации ДНК и репликации хроматина. Основную массу хроматина составляют белки гистоны. Циклин А/CDK2 индуцирует синтез гистонов, поставляя материал для упаковки новосинтезированной ДНК. К концу S фазы каждая хромосома состоит из двух пар идентичных сестринских хроматид. Каждая пара сестринских хроматид связана между собой комплексами белков, когезинами, которые регулируют процесс разделения сестринских хроматид при митозе [120].
G2 фаза. Прохождение клетки через G2 фазу и вход в митоз регулируется комплексом циклина В/CDK1 [121]. На сегодняшний день семейство циклинов В состоит из 5 белков – циклины В1, В2, В3, В4 и В5, из которых В1 является наиболее охарактеризованным [122]. Промотор циклина В1 содержит сайты связывания для множества транскрипционных факторов, в том числе B-Myb, NF-Y, USF, p53, p21, Ap-1, Ap-2, Ets-1 и др. [123]. Экспрессия гена циклина В1 наблюдается на протяжении всего клеточного цикла, однако в G2 уровень циклина В1 в 50 раз больше, чем в G1. Наивысшего уровня циклин В1 достигает в конце G2 фазы – начале митоза [124]. В середине митоза уровень циклина В1 резко снижается за счет работы АРС и остается низким до поздней S фазы, когда происходит ингибирование АРС с помощью Emi1 и комплекса циклин А/CDK2 [117, 125]. Усиление экспрессии циклина В1 приводит к его накоплению в цитоплазме, достигнув определенной концентрации циклин В связывается с CDK1. Образовавшийся комплекс циклин В1/CDK1 перемещается в ядро [126]. Транслокация циклин В1/CDK1 в ядро возможна только при фосфорилировании 5 серинов в сайте удерживающем циклин В1 в цитоплазме (cytoplasmic retention site, CRS): Ser116, Ser26, Ser128, Ser133 и Ser147 [126]. Показано, что ингибирование ERK1/2 препятствует фосфорилированию CRS, что приводит к удерживанию комплекса циклин В1/CDK1 в цитозоле [127]. Yang et al. предполагают, что CRS циклина В1 является частью последовательности для экспорта белка в ядро (nuclear export sequences, NES), с помощью которой циклин В1 взаимодействует с экспортирующим ядерным рецептором (chromosome region maintenance 1, CRM1) [128]. Однако комплекс остается неактивным, потому что протеинкиназы Wee1 и Myt1 фосфорилируют CDK1 по Thr14 и Tyr15, ингибируя активность CDK1. Wee1 находится в ядре, а Myt1 – на цитоплазматической стороне мембраны эндоплазматического ретикулума [129-131]. Интересно отметить, что активация ERK1/2 снижает активность Myt1 через активацию RSK, непосредственного субстрата ERK1/2, который фосфорилирует и ингибирует Myt1, приводя к остановке клеточного цикла в G2/M [132]. Для активации комплекса циклин В/CDK1 необходима фосфатаза Cdc25С, которая дефосфорилирует CDK1, убирая ингибирующие фосфаты [133, 134]. Активированный циклин В/CDK1 запускает события раннего митоза [129]. Параллельно с активацией циклин В/CDK1 происходит ингибирование Wee1 и Myt1. В течение интерфазы Cdc25C-associated protein kinase (C-TAK1) фосфорилируют Cdc25С по Ser216, что способствует его связыванию с белком 14-3-3 [135-137], который удерживает Cdc25С в цитоплазме, препятствуя перемещению фосфатазы в ядро и активации циклин В/CDK1 [138, 139]. Активирующаяся в ответ на повреждение ДНК Chk1 киназа тоже способна фосфорилировать Cdc25С, приводя к ингибированию фосфатазы и остановке прогрессии клеточного цикла [138, 140, 141].
Митоз. Основная задача митоза – равномерное распределение содержимого материнской клетки между двумя дочерними. Митоз состоит из нескольких подфаз (профаза, прометафаза, метафаза, анафаза, телофаза) и заканчивается цитокинезом, делением клетки надвое. События раннего митоза (профаза, прометафаза, метафаза) запускаются активированным во время G2 фазы комплексом циклин В/CDK1. Циклин В/CDK1 фосфорилирует субъединицы белка конденсина, это стимулирует свертывающую активность конденсина и инициирует конденсацию хромосом. Параллельно активируется сборка митотического веретена деления. Фосфорилирование белков, входящих в состав комплексов ядерных пор и ламины, приводит к разрушению ядерной оболочки. В конце метафазы циклин В/CDK1 активирует АРС. Клетки не входят в анафазу (не происходит разделения хроматид) до тех пор, пока все хромосомы не прикрепятся к веретену деления. Неприкрепленные хроматиды блокируют разделение хроматид. Это происходит, вероятно, за счет того, что неприкрепленные кинетохоры посылают сигнал АРС, блокирующий его активность [142]. Активность АРС регулируется с помощью двух дополнительных белков Cdc20 и Cdh1, которые выполняют функцию субстрат-специфических связывающих доменов. Циклин В/CDK1 фосфорилирует АРС, приводя к образованию комплекса АРС/Cdc20 и усилению активности АРС. АРС/Cdc20 ингибирует АРС/Cdh1 за счет фосфорилирования Cdh1. АРС/Cdc20 убиквитинирует белок секурин, что ведет к его деградации и активации протеолитического фермента, сепаразы, разрушающей комплексы когезинов, инициируя разделение хромосом и их расхождение к полюсам деления. Помимо участия в расхождении хромосом, АРС инициирует деградацию всех циклинов, приводя к инактивации всех CDK и дефосфорилированию их мишеней. Эти изменения необходимы для завершения митоза – разрушения веретена деления и цитокинеза. Деградация циклина В приводит к инактивации комплекса циклин В/CDK1, а, следовательно, и стимула активирующего АРС/Cdc20. Снижение АРС/Cdc20 в конце митоза способствует увеличению активности АРС/Cdh1. АРС/Cdh1 также как и АРС/Cdc20 убиквитинирует циклины приводя к их деградации [98]. В результате период времени от анафазы до поздней G1 является периодом инактивации CDK. Этот период необходим, так как сдерживает какое то время клетку от вступления в новый клеточный цикл. Некоторые клетки никогда не входят в клеточный цикл оставаясь в т.н. фазе покоя - G0, экспрессия генов циклинов и CDK в них подавлена [80].
Подводя итог вышесказанному, важно отметить следующее: пролиферация представляет собой последовательность переходов от G1 фазы клеточного цикла к S, G2 и митозу. События каждой фазы запускаются путем фосфорилирования определенных белков с помощью циклин-зависимых киназ. Активность циклин-зависимых киназ регулируется с помощью синтеза/деградации их функциональных субъединиц, циклинов, а также фосфорилирования/дефосфорилирования самих киназ. На сегодняшний день в литературе накоплено множество данных о взаимосвязи между состоянием редокс-статуса клетки и функционированием некоторых белков-регуляторов клеточного цикла, что позволяет предположить существование редокс-регуляции клеточного цикла.
1.2.2. Редокс-регуляция клеточного цикла
Впервые существование редокс-регуляции клеточного цикла было упомянуто в работе Rapkine. Он наблюдал колебания уровня растворимых в трихлоруксусной кислоте тиолов по мере прогрессии клеточного цикла. Концентрация тиолов падала в течение подготовки к делению и возрастала по мере формирования митотического аппарата [143]. Позже Kawamura наблюдал усиление окраски тиоловых групп (-SH) белков по мере сборки веретена деления в яйцах морского ежа находящихся в профазе. Окрашивание оставалось интенсивным в метафазе, постепенно снижалось по мере продвижения клеток по анафазе и было почти незаметным в телофазе [144]. Mauro et al., используя синхронизированные клетки HeLa, наблюдали снижение концентрации несвязанных с белками -SH групп в G1 фазе с последующим увеличением концентрации к концу S фазы. Концентрация восстановленных тиоловых групп в составе белков, наоборот, была максимальной в G1 и постепенно снижалась по мере прогрессии через S фазу [145]. Tu et al. продемонстрировали взаимосвязь между экспрессией генов и интенсивностью дыхательных процессов. Экспрессия генов, кодирующих рибосомальные белки, факторы инициации трансляции и других регуляторов синтеза белка, происходила во время фазы активного дыхания, что, возможно, связано с потреблением большого количества энергии во время синтеза белка. Репликация ДНК и прогрессия клеточного цикла, наоборот, происходили в условиях пониженной дыхательной активности, что, вероятно, способствовало снижению окислительного повреждения ДНК под действием АФК, образующихся в большом количестве в процессе дыхания [146]. Существование взаимосвязи между редокс-статусом и синтезом белка было подтверждено в работе Jonas et al. При исследовании синтеза ДНК по интенсивности включения синтетического аналога тимина - 5-бром-2-дезоксиуридина (БДУ) было обнаружено, что при окисленном состоянии редокс-статуса скорость синтеза ДНК минимальна и значительно возрастает при смещении редокс-статуса в более восстановленное состояние [147].
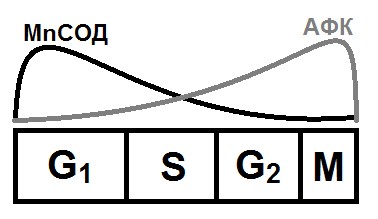
Рисунок 1.2. Изменения активности MnСОД и уровня АФК по мере прогрессии клеточного цикла.
Проведенные в нескольких независимых лабораториях исследования выявили, что по мере прогрессии клеточного цикла от G0/G1 к митозу происходит увеличение уровня АФК. Концентрация АФК, измеренная по флуоресценции редокс-чувствительного красителя DCFH2-DA, была максимальна в поздней S фазе, а также в G2+M по сравнению с G1 [26]. Параллельно с повышением АФК, увеличивался уровень основного редокс-буфера клетки – глутатиона (GSH) [148] – и достигал максимума в G2 и M фазах по сравнению с G1, что, вероятно, являлось ответной реакцией на рост АФК [47, 149]. Интересно отметить, что активность основного антиоксидантного фермента клетки - MnСОД – была максимальна в G1 фазе и постепенно снижалась по мере прогрессии через S и G2 фазы что, возможно, и являлось причиной роста АФК (см. Рис. 1.3) [27, 148, 150]. Более детальное исследование изменения уровня О2•- и Н2О2 по мере прогрессии клеточного цикла выявило, что О2•- повышен в G1 фазе, а уровень Н2О2 увеличивается в G2. Поскольку MnСОД играет важную роль в регуляции соотношение между О2•- и Н2О2, то было сделано предположение, что MnСОД выполняет роль переключателя пролиферативных процессов. Повышение активности MnСОД приводило к снижению уровня О2•-, увеличению уровня Н2О2 и усилению пролиферации. Снижение активности MnСОД сопровождалось ростом уровня О2•-, понижением уровня Н2О2 и замедлением пролиферации [27, 151]. Изменения внутриклеточного редокс-статуса при прогрессии клеточного цикла позволяют предположить существование т.н. редокс-цикла внутри клеточного цикла и тесно с ним взаимосвязанного. Причем, смещение редокс-статуса в более окисленное состояние сопровождается активацией пролиферации, а смещение редокс в более восстановленное состояние приводит к замедлению пролиферации [25, 152].
Молекулярные механизмы редокс-регуляции. С помощью комбинированного in vitro/биоинформатического анализа выявлено, что 92 из 1634 исследованных белков-регуляторов клеточного цикла содержат в своей структуре участки, способные модифицироваться в ответ на изменения редокс-статуса клетки. К редокс-чувствительным группам были отнесены тиольные группы цистеина и метионина (–SH), дисульфидные связи (-S-S-), ионы переходных металлов (Fe2+, Zn2+, Cu2+ и др), сопряженные системы связей в белках-переносчиках электронов дыхательной цепи (Рис. 1.3). Окисление тиоловых групп с образованием дисульфидных связей под действием Н2О2 часто сопряжено с внутримолекулярными конформационными изменениями, приводящими к активации/инактивации молекулы (Рис. 1.3. А, Б). Изменение степени окисления металлов-кофакторов киназ и фосфатаз под действием О2-• часто приводит к ослаблению связей, удерживающих металл в комплексе с белком, диссоциации металла из комплекса и нарушению редокс-регуляции (Рис. 1.3. В, Г) [29, 149].
Показано, что взаимодействие некоторых митогенов (EGF, PDGF и др.) с их рецепторами на поверхности мембран сопровождается образованием небольшого количества Н2О2 [153, 154]. Добавление Н2О2 в отсутствии EGF или PDGF индуцировало внутриклеточный сигнал сходный с сигналом факторов роста. Каталаза и пероксиредоксин ингибировали Н2О2–индуцированный сигналинг [154-156]. Показано, что присутствие Н2О2 необходимо для фосфорилирования и активации МАР-киназы ERK1/2 [155, 157]. Интересно отметить, что молекула малой ГТФазы Ras, активатора ERK1/2, содержит редокс-чувствительные цистеины [158]. Модификации цистеинов в молекуле Ras под действием Н2О2 может быть одним из механизмов редокс-регуляции ERK1/2 [159]. Ингибирование образования Н2О2 с помощью NAC или GSH, а также усиление экспрессии MnСОД препятствовало активации ERK1/2 и дальнейшей передачи сигнала от рецепторов факторов роста к ядру [28, 160].
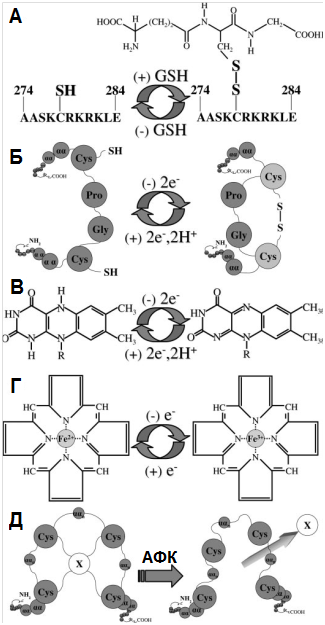
Рисунок 1.3. Примеры редокс-регулируемых модификаций в молекулах белков. Из [149] с изменениями. (А) Обратимое глутатионилирование остатка цистеина. (Б) Образование дисульфидной связи в молекуле тиоредоксина. (В) Взаимодействие электрона с изоаллоксазиновой сопряжённой циклической системой флавина – кофактора белка электронно-транспортной цепи. (Г) Окисленная иона железа в составе гемма. (Д) Диссоциация окисленного переходного металла (Х) из металлсвязывающего домена белка.
В нескольких независимых лабораториях было показано, что ДНК-связывающий домен некоторых факторов транксрипции (р53, NF-kB, AP-1, c-Myb, Ets, Sp-1, Egr-1 и др.) содержит консервативные цистеины, замещение или удаление которых приводит к нарушению редокс-регуляции и изменению активности/функции белка (см. [161] и ссылки в нем). Например, замещение консервативных цистеинов в молекуле опухолевого супрессора р53 приводит к снижению его ДНК-связывающих способностей. Неспособный связываться с ДНК белок не может выполнять свои функции фактора транскрипции и опухолевого супрессора, что часто приводит к трансформации клетки и чрезмерной пролиферации [162, 163]. Сходным образом замещение цистеинов в молекулах NF-kB и АР-1 и нарушение их редокс-регуляции способствует абберантной пролиферации и развитию опухолей [164, 165].
Показано, что промотор циклина D1, важного регулятора прогрессии клетки через G1 фазу, содержит сайты связывания редок-регулируемых факторов транскрипции, таких как NF-kB, AP-1, Sp-1 и др, то есть экспрессия циклина D1 зависит от состояния внутриклеточного редокс-статуса [166]. В условиях окислительного стресса происходит глутатионилирование c-Jun, одной из субъединиц АР-1, по Cys269, который находится в сайте связывания ДНК. Глутатионилирование ДНК-связывающего домена ингибирует активность АР-1 и репрессирует экспрессию циклина D1. В опухолевых клетках часто происходит мутация Cys269 и его замена на серин, что приводит к потере редокс-регуляции и абберантной пролиферации [167]. Чувствительность к состоянию редокс-статуса была также показана для c-Myb и NF-Y, регулирующим транскрипцию циклинов В1 и А. Фактор транскрипции b-Myb содержит семь редокс-чувствительных цистеинов. Смещение окислительно-восстановительного равновесия в более окисленное состояние с помощью окислителя диамида значительно снижало активность b-Myb, приводя к нарушению связывания транскрипционного фактора с ДНК. Интересно отметить, что повышенный уровень b-Myb часто встречается в опухолевых клетках. Как отмечалось выше, большинство опухолевых клеток характеризуется повышенным содержанием АФК, что возможно и приводит к нарушению регуляции пролиферации с помощью b-myb [168]. Фактор транскрипции NF-Y на 30% существует в виде неактивных димеров. Добавление восстановителя, дитиотреитола, к NF-Y способствовало усилению связывания NF-Y с ДНК. Замещение Cys85 и Cys89 в молекуле NF-Y на серины приводило к тому, что NF-Y присутствовал только в форме мономеров и его активность не зависела от дитиотреитола [169].
![3. Редокс-регуляция клеточного цикла. Из [152] с изменениями, см.-3](/images1/370825/3-redoks-regulyaciya-kletochnogo-cikl.png)
Рисунок 1.3. Редокс-регуляция клеточного цикла. Из [152] с изменениями, см. пояснения в тексте.
Повышение уровня GSH при обработке мышиных фибробластов, находящихся в ранней G1 фазе, с помощью NAC приводило к нарушению прогрессии клеточного цикла, увеличению количества клеток в G1 фазе и снижению количества клеток в S фазе. Нарушению прогрессии клеточного цикла предшествовало уменьшение уровня циклина D1, увеличение уровня р27, ингибитора Cdk4/6, и дефосфорилирование рRb, что указывало на зависимость уровня циклина D1, р27 и рRb от состояния редокс-статуса. После удаления NAC из среды наблюдали кратковременный скачок прооксидантного уровня (Н2О2) и возобновление прогрессии клеточного цикла [170]. Полученные данные указывали на необходимость присутствия небольших количеств Н2О2 для перехода клеток из G1 в S фазу клеточного цикла.
Подобно действию NAC, усиление экспрессии каталазы или GPx, катализаторов разложения Н2О2, приводило к снижению внутриклеточного уровня Н2О2 и остановке клеточного цикла в G1 фазе [63, 171, 172]. Обработка Her1 фибробластов с помощью Н2О2, наоборот, повышала уровень циклина D1, что предположительно было результатом ингибирования деградации циклина D1 [173]. Несмотря на то, что повышенный уровень GSH ингибировал прогрессию клеточного цикла, полное удаление тиолов из среды также препятствовало переходу клеток в S фазу. Удаление тиолов из среды культивирования лимфоцитов, препятствовало экспрессии и фосфорилированию рRb под действием IL-2. Добавление GSH, NAC и 2-меркаптоэтанола обращало этот эффект [174]. Из полученных данных можно сделать вывод о том, что хотя небольшое увеличение уровня АФК необходимо для активации рRb, чрезмерное повышение прооксидантного уровня приводит к нарушению клеточного цикла.
Дальнейшее исследование роли GSH и АФК в регуляции клеточного цикла выявило, что снижению экспрессии циклина D1 в клетках, обработанных NAC, предшествовало повышение уровня О2•-. Инкубация клеток с Tiron, ловушкой для О2•-, препятствовало нарушению прогрессии клеточного цикла под действием NAC, указывая на то, что О2•- может оказывать ингибирующее действие на экспрессию циклина D1 [175]. Однако, снижение уровня О2•- с помощью темпола, препятствовало накоплению циклина А, регулятора G1/S перехода, и прогрессии через S фазу, приводя к остановке клеточного цикла в поздней G1 фазе. Удаление темпола из среды способствовало восстановлению прогрессии клеточного цикла. Эти данные указывают на необходимость присутствия О2•- для перехода клетки из G1 в S фазу. Повышенная экспрессия Emi1, ингибитора комплекса APC/Cdh1, отменяла эффект темпола. Клетки экспрессирующие Emi1, демонстрировали уровень циклина А сравнимый с контролем и отсутствие блока в G1 фазе. К сходным результатам приводило применение ингибиторов 26S протеасомы. Как было отмечено выше убиквитинирование циклина А с помощью комплекса APC/Cdh1 в течение G1 фазы препятствует переходу в S фазу. Исходя из имеющихся данных можно предположить, что О2•- играет важную роль в ингибировании APC/Cdh1 и поэтому его присутствие необходимо для перехода в S фазу. Таким образом, было показано, что повышение уровня АФК при прогрессии из G1 в S необходимо для ингибирования деградации циклина А под действием АРС [176]. Отсюда следует, что присутствие О2•- в начале G1 фазы ингибирует экспрессию циклина D1 и приводит к остановке клеточного цикла. Однако в конце G1 фазы, О2•- необходим для прогрессии из G1 в S. Кажущаяся несогласованность является лишь еще одним доказательством существования редокс-цикла внутри клеточного цикла, а также подчеркивает сложность и важность редокс-модификаций для регуляции пролиферации.
Еще один фермент S фазы, топоизомераза II, играет важную роль в поддержании целостности ДНК, путем распутывания переплетенных в процессе репликации сестринских хроматид и способствуя релаксации ДНК. Goswami et al показали, что уровень мРНК топоизомеразы варьирует в зависимости от фазы клеточного цикла достигая максимума в S фазе. Дальнейшие исследования выявили, что экспрессия топоизомеразы II регулируется за счет взаимодействия 3`-UTR последовательности мРНК с редокс-чувствительными белками [32, 57].
Фосфатаза Cdc25С, важный регулятор прогрессии клеточного цикла через G2 фазу, содержит в активном центре цистеин (Cys377) и поэтому чувствителен к изменениям редокс-статуса. Образование связи между Cys377 и Cys330 приводит к усилению связывания Cdc25С с белком 14-3-3, что препятствует перемещению Cdc25С в ядро и активации комплекса циклин В1/Cdk1 (см. Рис. 1.4) [138, 177]. В независимом исследовании было показано, что обработка клеток Н2О2 приводит к инактивации Cdc25C, в то время как белок мутантный по Cys377 и Cys330 был нечувствителен к действию Н2О2. [177, 178]. Как было отмечено выше, уровень GSH возрастает по мере приближения к митозу [148], это вероятно способствует переходу Cdc25C в более стабильное состояние и активации циклин В/Cdk1.
![4. Редокс-регуляция активности Cdc25C фосфатазы. Из [177] c-4](/images1/370825/4-redoks-regulyaciya-aktivnosti-cdc2.jpg)
Рисунок 1.4. Редокс-регуляция активности Cdc25C фосфатазы. Из [177] c изменениями. См. пояснения в тескте.
Chang et al. показали, что в G2 фазе активная CDK1 фосфорилирует пероксиредоксин I, внутриклеточный фермент катализирующий деградацию Н2О2, ингибируя его активность. Накопление Н2О2, в результате снижения активности пероксиредоксина I, представляется необходимым для прогрессии через G2 фазу в М [179]. Как было отмечено выше, активность MnСОД снижается по мере прогрессии клеточного цикла от G1 к М фазе, способствуя повышению уровня АФК. Повышение экспрессии MnСОД в клетках карцином усиливало формирование индуцированного радиацией G2 блока, указывая на то, что высокий уровень MnСОД препятствует прогрессии клеток через G2 фазу [180]. Другой внутриклеточный антиоксидант, витамин С, также был способен индуцировать G2 блок в клетках карциномы HeLa и глиобластомы T98G человека. Индукция G2 блока под действием витамина С сопровождалась усилением экспрессии циклина В1 и снижением уровня активного Cdc25С, активатора комплекса циклин В1/CDK1 [181].
Присутствие редокс-чувствительных цистеинов в молекулах убиквитин-активирующего (Е1) и убиквитин-коньюгирующего (Е2) предполагают существование редокс-регуляции процессов деградации белка. Повышение уровня восстановленного глутатиона (GSH) ингибировало активность Е1 и Е2 [182], а также 20S субъединицу протеасомы, препятствуя протеолизу белка [183]. Н2О2, наоборот, стимулировал процессы убиквитинирования, за счет усиления экспрессии E2 и убиквитин-лигазы (Е3) [184].
Резюмируя сказанное выше, важно подчеркнуть следующее: прогрессия клеточного цикла сопровождается не только изменениями уровня циклинов и активности CDK, но и колебаниями соотношения про- и антиоксидантов внутри клетки (окислительно-восстановительного баланса). Исходя из этого, было предположено существование т.н. редокс-цикла внутри клеточного цикла и тесно с ним взаимосвязанного. Пертурбации внутриклеточного окислительно-восстановительного баланса ведут к нарушениям прогрессии клеточного цикла, указывая на возможность регуляции пролиферации через модуляцию редокс-статуса клетки.
РАЗДЕЛ 1.3. Современное состояние знаний о противоопухолевом эффекте короткоцепочечных пептидов
1.3.1. Карнозин и его производные. Общая характеристика
Карнозин представляет собой природный короткоцепочечный пептид, состоящий из двух аминокислот – -аланина и L-гистидина. В организме человека карнозин представлен во многих тканях (мышцы, мозг, печень, ткани глаза, желудок, почки, легочная ткань), достигая наиболее высокой концентрации в скелетных мышцах (~ 20 мM) и в обонятельных луковицах головного мозга (~ 2.5 мM) [1, 2].
Карнозин синтезируется с помощью фермента карнозин-синтетазы [3-6], которая катализирует образование пептидной связи между -аланином и L-гистидином, используя энергию АТФ и ионы Mg2+:
-аланин + L-гистидин + АТФ = карнозин + Pi + АДФ [7]
Карнозин-синтетаза относится к семейству ATP-grasp ферментов, катализирующих АТФ-зависимое присоединение карбоксила одной молекулы к амино- или тиоловой группе другой с образованием АДФ и органического фосфата (Pi) [185]. Ген карнозин-синтетазы располагается в 11q13 хромосоме человека и экспрессируется главным образом в мозге и мышцах [7]. Bauer et al. показали, что карнозин и его производные синтезируется в основном клетками глии. Интересно, что опухолевые клетки, глиомы С6, тоже активно синтезировали карнозин и родственные соединения [186]. Синтез карнозина в нейронах обнаружен не был, однако нейроны были способны поглощать карнозин из среды и метаболизировать его [187-190]. Интересно отметить, что усиление синтеза карнозина сопровождалось дифференцировкой глиальных клеток, что оценивалось по усилению иммуноокрашивания антителами к маркерам дифференциации [189]. Сходный эффект наблюдали при обработке скелетных мышц новорожденных цыплят. Усиление синтеза карнозина замедляло скорость деления клеток и способствовало превращению миобластов в миотубулы [191, 192].
В организме человека существует два фермента, способных расщеплять карнозин на составные аминокислоты: сывороточная (CN1) [193] и тканевая/цитозольная (CN2) карнозиназы [194, 195]. CN1 экспрессируется главным образом в мозге, и в меньшей степени в печени. Иммуногистохимическое исследование выявило присутствие CN1 в цитозоле пирамидальных нейронов гиппокампа, а также в больших и малых нейронах коры больших полушарий. Цитозольная фракция клеток, секретирующих CN1, не деградировала карнозин, указывая на то, что CN1 относится к секретируемым белкам и активен только после секреции [9]. В отличие от CN1, CN2 экспрессируется повсеместно в тканях человека и является цитозольным ферментом. Больше всего этого фермента содержится в почках и печени, в меньшем количестве обнаружен в мозге, селезенке и поджелудочной железе, следовые количества – в легких, семенниках и яичниках, отсутствует в сердце [9]. Ферменты характеризуются разными рН-оптимумами работы. CN1 демонстрирует наибольшую активность при рН 7,5 – 8,5, CN2 характеризуется более узким оптимумом работы рН с максимумом около рН 9,5. Таким образом, при физиологических pH карнозин гидролизуется преимущественно с помощью CN1 [9]. Интересно отметить, что экспрессия CN1 отсутствует в пренатальном периоде и у новорожденных, что указывает на усиление экспрессии CN1 с возрастом [9, 196]. Кроме того, у пожилых людей обнаружен более низкий уровень гомокарнозина в сыворотке крови по сравнению с молодым поколением [197].
У карнозина существует несколько природных производных, в том числе: N-ацетил-карнозин (N-ацетил--аланил-L-гистидин), анзерин (-аланил-1-метил-L-гистидин) и гомокарнозин (у-амино-бутирил-L-гистидин) (см. Рис. 1.5). Считается, что -аланил-содержащие дипептиды преимущественно располагаются в скелетных мышцах, в то время как дипептиды содержащие -амино-масленную кислоту (ГАМК) типичны для центральной нервной системы [7]. Ацетилированная форма карнозина присутствует в сердечной мышце и в мозге [198]. Предполагается, что образование ацетилкарнозина происходит за счет ацетилирования карнозина, однако информация о ферментах, катализирующих данную реакцию пока отсутствует. Гомокарнозин (0,3-1,5 мМ [199]) синтезируется в мозге из ГАМК и L-гистидина с помощью карнозин-синтетазы. Однако скорость синтеза гомокарнозина при прочих равных условиях гораздо медленнее, чем для карнозина [5, 7]. Синтез анзерина (до 40 мМ в мышцах птиц [200]) происходит в мышцах позвоночных животных путем метилирования карнозина с помощью метилтрансферазы [201]. Анзерин не обнаружен в организме человека. Расщепление производных карнозина осуществляет сывороточная карнозиназа (CN1), при этом скорость гидролиза гомокарнозина составляет 11% от скорости гидролиза карнозина или анзерина [202].
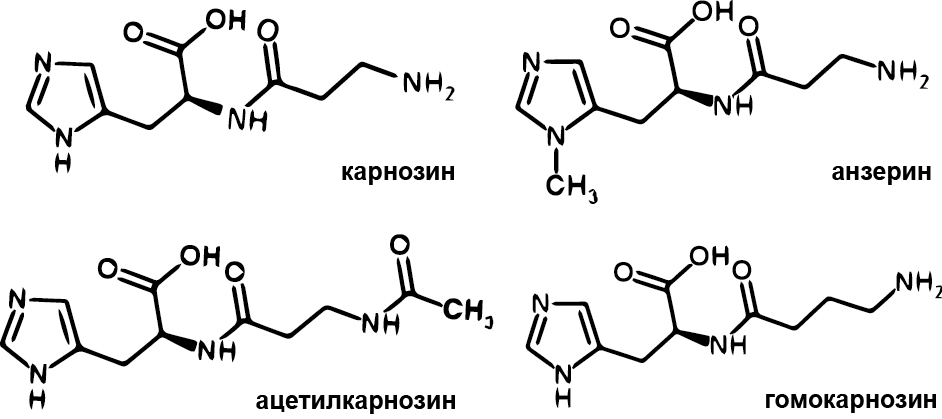
Рисунок 1.5. Структура карнозина и его производных. Из [203] изменениями.
В организме человека карнозин выполняет множество функций, среди которых: pH-буфер в мышцах [10], хелатор металлов переменной валентности (Fe, Cu, Zn, Co) [11-13, 204, 205], нейротрансмиттер в обонятельных луковицах [2, 6], антиоксидант [15-18, 206, 207], ингибитор опухолевого роста [20, 21, 23, 208-210].
1.3.2. Антиоксидантные свойства карнозина и его производных
Карнозин эффективно препятствовал окислению 2,5-бис(гидроксиметил)фурана (BHMF) и N,N-диметил-4-нитрозоанилин (RNO) под действием света в растворе, содержащем фотосенсибилизатор [211, 212]. BHMF и RNO представляют собой ловушки синглетного кислорода (1О2), ингибирование окисления ловушек с помощью карнозина указывает на способность карнозина взаимодействовать с 1О2. Кроме того, карнозин снижал интенсивность хемилюминесценции, индуцируемой синглетным кислородом, образующимся в смеси Н2О2+NaClO, подтверждая способность тушить 1О2 [213]. Интересно отметить, что при эквимолярных концентрациях эффективность тушения 1О2 карнозином была выше, чем у гистидина, известной ловушки для 1О2 [211, 212, 214]. Более высокая эффективность карнозина по сравнению с гистидином, возможно, связана с особенностями внутримолекулярных взаимодействий (влияние -аланина), что приводит к большей реакционной способности гистидина в молекуле карнозина.
Методом синхронной регистрации светопоглощения при радиолизе водного раствора карнозина было выявлено образование комплекса между супероксидом и карнозином с частичным переносом заряда. Образование комплекса приводило к снижению константы скорости дисмутации О2-• и повышению его стабильности в водном растворе [14]. Способность карнозина взаимодействовать с супероксид анионом была подтверждена в независимой работе Klebanov et al, где карнозин препятствовал восстановлению нитро-голубого тетразолия под действием О2-• образованного в смеси ксантин+ксантиноксидаза [17]. Сравнение эффективности тушения О2-•, образующегося в смеси глутатион+пероксидаза+люминол, выявило, что карнозин и гистидин демонстрировали примерно одинаковую эффективность, в то время как -аланин был неактивен, что указывает на определяющую роль гистидина при взаимодействии карнозина с супероксидом [215].
С помощью метода электронного парамагнитного резонанса (ЭПР) было продемонстрировано, что добавление карнозина к смеси реакции Фентона приводит к формированию продукта способного конкурировать со спиновой ловушкой для OH• - 5,5-диметилпирролин-N-оксидом (DMPO) [16, 216]. Гистидин и гомокарнозин взаимодействовали с OH• менее эффективно, чем карнозин. Образование комплексов OH• с -аланином и ГАМК обнаружено не было. Интересно отметить, что эффективность связывания OH• в смеси -аланин+гистидин была ниже, чем в присутствии карнозина, что еще раз подтверждает важную роль внутримолекулярных взаимодействий для антиоксидантного эффекта карнозина [16].
В ряде работ были продемонстрированы способности карнозина взаимодействовать с продуктами ПОЛ, препятствуя распространению окислительного повреждения. Карнозин сдерживал накопление малонового альдегида, образующегося в результате окисления фрагментов саркоплазматического ретикулума, и способствовал сохранению функций мембран [18]. Карнозин также снижал количество пероксильных радикалов, образующихся в липосомах печени в результате инициации ПОЛ с помощью азосоединений [15]. С использованием внеклеточных систем Zhou и Decker наглядно продемонстрировали взаимодействие карнозина с ненасыщенными альдегидами (транс-2-гексеналь, транс-2-ноненаль), а также 4-гидроксиноненалем – опасным продуктом повреждающим ДНК [19, 217]. Гистидин и имидазол также демонстрировали защитное действие, в то время как -аланин и ГАМК не оказывали какого либо эффекта [15, 19].
Одним из проявлений антиоксидантных свойств карнозина является его радиомодифицирующий эффект. Повреждающее действие ионизирующего излучения связано, главным образом, с развитием мощнейшего окислительного стресса в результате радиолиза воды. Пероральное введение мышам карнозина в течение 20 суток перед облучением способствовало повышению выживаемости животных [218]. Радиомодифицирующее действие карнозина было подтверждено с помощью метода эндогенных селезеночных колоний. Было обнаружено увеличение эффективности образования колоний гематопоэтическими стволовыми клетками из костного мозга мышей, которым перед облучением вводили карнозин, по сравнению с контрольными животными [219, 220].
1.3.3. Эффект карнозина и его производных на пролиферацию нормальных и опухолевых клеток
Продолжительное культивирование нормальных человеческих фибробластов с карнозином (10 - 50 мМ) приводило к удлинению продолжительности жизни клеток, что выражалось в увеличении количества популяционных удвоений, т.н. предела Хейфлика [221]. Кроме того, отмечали задержку развития старческого фенотипа в виде усиления зернистости и потери вытянутой фибробластоподобной формы [22]. Добавление карнозина к клеткам долгое время культивируемым на среде без карнозина приводило к обращению старческого фенотипа и способствовало увеличению продолжительности их жизни в сравнении с контролем. Удаление карнозина из среды нивелировало описанный эффект [222]. Карнозин также способствовал увеличению эффективности посева фибробластов. При посеве клеток в низких разведениях, в чашках инкубируемых с карнозином образовывалось больше колоний, чем в контрольных, что указывает на активацию пролиферативных процессов фибробластов под действием карнозина [222]. Позже Shao et al. продемонстрировали замедление скорости укорачивания теломер и снижение количества дефектов в ДНК теломер под действием карнозина [223]. Укорачивание теломер представляется одним из основных факторов ограничивающий количество клеточных делений [224], замедление этого процесса может быть одним из механизмов удлинения предела Хейфлика и задержки формирования старческого фенотипа с помощью карнозина.
В 1986 году, японские ученые Nagai и Suda впервые описали эффект карнозина на опухолевый клетки, который в корне отличался от действия карнозина на нормальные клетки. Подкожные инъекции карнозина мышам линии ddY, которым предварительно вживляли клетки саркомы, способствовали снижению интенсивности пролиферации опухолевых клеток и увеличивали выживаемость животных [20]. В независимом исследовании Renner et al. на мышах с ксенографами HER2/neu NIH3T3 фибробластов показали, что ежедневные внутрибрюшинные инъекции карнозина существенно подавляют пролиферацию злокачественных клеток и опухолевый рост. Снижение скорости пролиферации опухолевых клеток под действием карнозина сопровождалось снижением синтеза АФК и активности лактатдегидрогеназы (ЛДГ). Кроме того, в опухолях мышей, которым делали инъекции карнозина карнозина было обнаружено меньшее количество митозов, чем у контрольных животных [21]. Сходные изменения наблюдали в первичной культуре клеток мультиформной глиобластомы (Glioblastoma multiforme). Обработка клеток карнозином (20-50 мМ) в течение 4 дней приводила к уменьшению внутриклеточного уровня АТФ и ЛДГ, а также к снижению синтеза ДНК [208]. Ингибирование митохондриального дыхания с помощью KCN в клетках обработанных карнозином не влияло на выработку АТФ или выживаемость клеток. Обработка клеток глиобластомы оксаматом – ингибитором ЛДГ – приводила к резкому снижению концентрации АТФ, при этом карнозин приводил к дальнейшему снижению концентрации АТФ. Исходя из полученных данных авторами был сделан вывод о том, что карнозин индуцирует изменения на уровне гликолиза, которые приводят к ингибированию клеточной пролиферации [225]. Holliday и McFarland наглядно продемонстрировали избирательность ингибирующего эффекта карнозина на рост опухолевых клеток [23, 226]. Добавление карнозина к смеси опухолевых клеток (HeLa) и нормальных человеческих фибробластов (MRC-5) приводило к выборочному уничтожению опухолевых HeLa клеток и способствовало удлинению продолжительности жизни фибробластов [23]. Интересно отметить, что действие карнозина на две активно пролиферирующие культуры – эмбриональные стволовые (ЭС) клетки мыши и иммортальные клетки эмбриональной терато-карциномы (ЭК) мыши также было различно. Карнозин препятствовал росту злокачественных ЭК-клеток, не затрагивая роста ЭС-клеток [226]. Полученные данные указывали на то, что ни степень дифференцировки, ни скорость пролиферации клеток не являются определяющими факторами избирательного действия карнозина на опухолевые клетки.
Количественный анализ экспрессии белков в клетках глиобластомы в контроле и после обработки карнозином выявил изменения в экспрессии 31 белка под действием карнозина, в том числе: Von Hippel–Lindau binding protein 1 (VBP1), BCL2-associated athanogene 2 (BAG2), GrpE-like protein chaperone (GrpEL), transaldolase 1 (TALDO), uroporphyrinogen decarboxylase (UROD) и Obg-like ATPase 1 (OLA1) [227]. Интересно отметить, что VBP1, BAG2 и GrpEL участвуют в регуляции активности Hypoxia-inducible factor 1 (HIF-1) [227-230], а BAG2 и GrpEL вовлечены в регуляцию активности белков теплового шока (Heat shock protein 70, Hsp70) [231]. Оверэкспрессия HIF-1 и Hsp70 часто способствует злокачественной трансформации [232, 233]. Приведенные данные указывают на возможное участие регуляторных каскадов HIF-1 и Hsp70 в антипролиферативном эффекте карнозина.
В нашей лаборатории было показано, что карнозин замедляет рост уровня фосфо-ERK1/2 и JNK в гранулярных клетках мозжечка в условиях окислительного стресса, индуцированного активацией глутаматных рецепторов [234]. Как было отмечено выше ERK1/2 представляет собой редокс-чувствительную МАР-киназу, регулирующую множество внутриклеточных функций, в том числе и пролиферативные процессы. Было показано, что ингибирование образования Н2О2 препятствует активации ERK1/2 и развитию пролиферативного ответа [235]. Таким образом, понижение уровня АФК под действием карнозина представляется одним из механизмов подавления активации ЕRK1/2 [234, 236]. Повышенный уровень ERK1/2 часто встречается при онкологических заболеваниях [105]. Ингибирование роста ERK1/2 может быть одним из механизмов антипролиферативного эффекта карнозина.
Исследование противоопухолевого эффекта производных карнозина (анзерина, гомокарнозина и D-карнозина) выявило, что только анзерин способен ингибировать рост трансформированных клеток. Также было обнаружено, при равных концентрациях (20 мМ) -аланин не оказывал какого либо влияния на опухолевые клетки, в то время как гистидин убивал все клетки (нормальные и опухолевые) [23]. Полученные данные явно указывают на определяющую роль гистидина в проявлении противоопухолевого эффекта карнозина.
1.3.4. Пинеалон. Общая характеристика и свойства
В настоящее время установлена роль регуляторных короткоцепочечных пептидов в формировании адаптационного ответа организма на стресс и нарушения гомеостаза [237]. Будучи эндогенными компонентами живой клетки, пептидные биорегуляторы обладают разнообразным биологическим действием, они эффективны в низких дозах, не вызывают побочных эффектов [238]. Однако их терапевтическое применение ограничено проницаемостью через гемато-энцефалический барьер (ГЭБ) и относительно быстрой скоростью распада. Преодолеть эти проблемы можно путем синтеза короткоцепочечных аналогов нейропептидов, сохраняющих их специфическую активность [239-243].
Пинеалон представляет собой синтетический трипептид состоящий из трех аминокислот глутаминовой кислоты, аспарагиновой кислоты и аргинина (Glu-Asp-Arg), синтезированный на основе анализа экстракта коры головного мозга крупного рогатого скота (Рис. 1.6). Последовательность Glu-Asp-Arg была выбрана как наиболее часто встречающаяся в "активных участках" наиболее функционально-значимых в своей группе полипептидов, содержащихся в животных экстрактах.
Пинеалон демонстрирует эффективные антиоксидантные свойства. Пинеалон усиливал устойчивость экспериментальных животных к пренатальной и острой сублетальной гипобарической гипоксии. Введение пинеалона крысам чувствительным к гипоксии усиливало активность антиоксидантных ферментов – СОД и GPx. В культуре гранулярных клеток мозжечка пинеалон снижал уровень АФК и клеточную гибель, индуцированные активацией глутаматных рецепторов NMDA-класса [244, 245]. На модели пренатальной гипергомоцистеинемии было показано, что инъекции пинеалона самкам улучшают когнитивные способности потомства и способствуют развитию устойчивости к окислительному стрессу [246]. Кроме того, пинеалон препятствовал увеличению уровня АФК в гранулярных клетках мозжечка крысы под действием гомоцистеина (ГЦ) и затормаживал активацию ERK1/2 [247]. Недавно были получены данные о возможном участии пинеалона в эпигенетической регуляции. Пинеалон проникал в ядро клетки, а также взаимодействовал с некоторыми промоторными последовательностями, в частности с одноцепочечной oligo(dGC). Интересно, что предпочтительным местом связывания пинеалона была последовательность CNG, которая также является местом метилирования ДНК [248, 249]. Исходя из способностей пинеалона модулировать внутриклеточный уровень АФК и связываться с ДНК можно предположить, что пинеалон также способен участвовать в регуляции пролиферации.
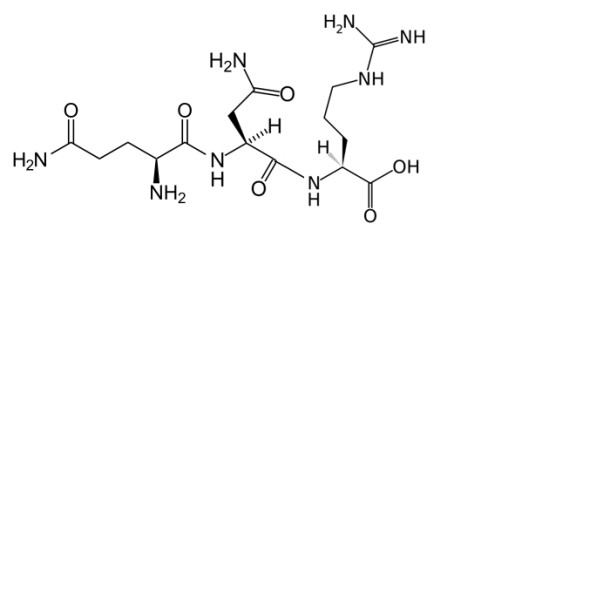
Рисунок 1.6. Химическая структура молекулы пинеалона.
Подводя итог всему выше сказанному, можно заключить, что антипролиферативный эффект карнозина известен давно, однако его молекулярный механизм до сих пор непонятен. Понимание механизма действия карнозина помогло бы расширить область применения природного дипептида карнозин на практике, например в терапии опухолевых заболеваний. На сегодняшний день накоплено множество сведений об участии про- и антиоксидантов в регуляции процессов клеточной пролиферации. Поскольку, карнозин демонстрирует эффективные антиоксидантные свойства, было сделано предположение о том, что ингибирующий эффект карнозина на пролиферацию опухолевых клеток является следствием его антиоксидантной активности. Сравнительный анализ действия карнозина и его производных на пролиферацию опухолевых клеток помог бы определить функциональную нагрузку отдельных частей молекулы при антипролиферативном эффекте и пути улучшения эффективности антипролиферативных свойств карнозина.
ГЛАВА 2. МЕТОДЫ ИССЛЕДОВАНИЯ
2.1. Культуры клеток
В работе были использованы следующие клеточные линии: феохромоцитома крысы (РС-12); карцинома горла и рта (FaDu, Cal27) и молочной железы (MB231) человека; глиобластома человека (U-118-MG). Клетки PC-12 культивировали в среде RPMI 1640 с 25 мМ HEPES и 24 мМ бикарбоната натрия (ПанЭко, Россия), с добавлением 2 мМ глутамина (ПанЭко, Россия), 20 мкг/мл гентамицина (ПанЭко, Россия) и 10% эмбриональной телячьей сыворотки (ПанЭко, Россия). Клетки FaDu и Cal27 культивировали в среде DMEM (Gibco, США) с добавлением 10% эмбриональной телячьей сыворотки (HyClone, США) и 100 ед/мл ПенСтреп (Gibco, США). Клетки MB231 инкубировали в среде RPMI 1640 (Gibco, США) с добавлением 10% эмбриональной телячьей сыворотки (HyClone, США) и 100 ед/мл ПенСтреп (Gibco, США). Клетки U-118-MG культивировали в смеси DMEM:F12 (1:1) (Gibco, США) с добавлением 15 мМ HEPES (Gibco, США), 1 мМ пирувата натрия (Gibco, США), 10 мкг/мл инсулина (Sigma Aldrich, США), 5 нг/мл фактора роста фибробластов (Sigma Aldrich, США), 15% эмбриональной телячьей сыворотки (HyClone, США) и 100 ед/мл ПенСтреп (Gibco, США). Клетки содержали в СО2-инкубаторе (ShelLab, США) в присутствии 5% СО2, 98% влажности и 37С.
2.2. Исследуемые соединения
Карнозин (чистота 99%) был любезно предоставлен «Hamari Chem., Ltd», Осака, Япония. Гомокарнозин (чистота 99%) и анзерин (чистота 99%) были приобретены, соответственно, в «Hamari Chem., Ltd» и «Yaizu Suisankagaku Industry Co.,Ltd.» (чистота 99%). N-ацетилкарнозин был синтезирован рутинным путем в Лаборатории клинической и экспериментальной нейрохимии ФГБУ «НЦН» РАМН (чистота 98,7%, определена с помощью HPLC). L-гистидин--аланин был синтезирован рутинным путем в Исследовательском Институте Химического Разнообразия (чистота 98%, определена с помощью HPLC). Пинеалон был предоставлен Научно-производственным центром ревитализации и здоровья.
2.3. Исследование клеточной пролиферации
2.3.1. Время удвоения популяции
Время удвоения популяции (Тd) – это период времени, за который происходит увеличение количества клеток в два раза. Для вычисления Тd клетки высаживали в чашки Петри в небольших разведениях. Каждые два дня клетки снимали с помощью трипсина (Gibco, США), считали их количество с помощью Z1 Coulter Counter (Beckman Coulter, США) и строили кривые роста (Рис. 2.1). Время удвоения популяции (Тd) рассчитывали на экспоненциальном участке роста по формуле:
Тd=0.693t/ln(Nt/N0),