

Электрохимические процессы галогенирования и окисления пространственно-затрудненных аминов и нитроксильных радикалов ряда 2,2,6,6-тетраметилпиперидина и их практическое использование
На правах рукописи
Жукова Ирина Юрьевна
ЭЛЕКТРОХИМИЧЕСКИЕ ПРОЦЕССЫ ГАЛОГЕНИРОВАНИЯ И ОКИСЛЕНИЯ ПРОСТРАНСТВЕННО-ЗАТРУДНЕННЫХ АМИНОВ И НИТРОКСИЛЬНЫХ РАДИКАЛОВ РЯДА 2,2,6,6-ТЕТРАМЕТИЛПИПЕРИДИНА И ИХ ПРАКТИЧЕСКОЕ ИСПОЛЬЗОВАНИЕ
05.17.03-«Технология электрохимических процессов и защита от коррозии»
АВТОРЕФЕРАТ
диссертации на соискание ученой степени
доктора технических наук
Новочеркасск – 2010
Работа выполнена в ГОУ ВПО «Южно-Российский государственный технический университет (Новочеркасский политехнический институт)» на кафедре «Химическая технология высокомолекулярных соединений, органическая, физическая и коллоидная химия».
Научный консультант: доктор химических наук, профессор
Каган Ефим Шоломович
Официальные оппоненты: доктор химических наук, профессор
Берберова Надежда Титовна;
доктор химических наук, профессор
Гультяй Вадим Павлович;
доктор технических наук, профессор
Селиванов Валентин Николаевич
Ведущая организация: Южный федеральный университет,
(г. Ростов-на-Дону)
Защита состоится « » июня 2010 г. в 11.00 часов, в ауд. 107 на заседании диссертационного совета Д 212.304.05 при Южно-Российском государственном техническом университете (Новочеркасском политехническом институте) по адресу: 346428, Ростовская обл., г. Новочеркасск, ул. Просвещения, 132.
С диссертацией можно ознакомиться в библиотеке Южно-Российского государственного технического университета (НПИ).
Автореферат разослан « » мая 2010 г.
И.о. ученого секретаря
диссертационного совета Савостьянов А.П.
Общая характеристика работы
Актуальность работы.
Открытие в начале 60-х годов прошлого века нитроксильных радикалов (НР) ряда 2,2,6,6-тетраметилпиперидина (2,2,6,6-ТМП) и реакций, происходящих с сохранением радикального центра, вызвало большой интерес к методам синтеза, свойствам и практическому использованию НР. Важнейшими результатами исследований в этой области является открытие нового класса стабилизаторов полимеров против термо- и фотодеструкции на основе производных 2,2,6,6-ТМП и метода спиновых меток. В последние годы наиболее активно изучаются окислительно-восстановительные свойства НР ряда 2,2,6,6-ТМП, так как эти радикалы образуют уникальную обратимую окислительно-восстановительную систему НРоксоаммониевая соль, которая используется в процессах окисления органических веществ, а также в качестве положительного электрода органических радикальных аккумуляторов. Об интересе к этой области свидетельствуют многочисленные публикации и обзоры, посвященные химии и применению НР.
Несмотря на то, что НР интенсивно изучаются более 50-ти лет, многие проблемы остаются нерешенными. В частности, практически не изучены реакции электрохимического окисления ПЗА ряда 2,2,6,6-ТМП, которые могут стать важными для синтеза НР и понимания процессов, происходящих при стабилизации полимеров. Окисление ПЗА является основным методом синтеза НР ряда 2,2,6,6-ТМП. Кроме этого, в основе применения ПЗА в качестве стабилизаторов полимеров лежит реакция их окисления, приводящая к образованию аминильных радикалов и обрыву цепей окисления полимеров.
Важную роль в процессах синтеза ПЗА ряда 2,2,5,5-тетраметилпирролидина и НР этого ряда, широко применяемых в научных исследованиях в качестве спиновых меток для изучения биополимеров, играют реакции галогенирования 4-оксо-2,2,6,6-тетраметилпиперидина (триацетонамина, ТАА). Синтез веществ этого ряда основан на предварительном получении галогенпроизводных ТАА. Методы синтеза этих соединений являются многостадийными и сложными. Электрохимические методы синтеза ПЗА ряда 2,2,5,5-тетраметилпирролидина, которые могут быть более эффективными, чем известные способы их получения, практически не изучены.
Большой интерес для применения в органическом синтезе в качестве реагентов представляют N-галогенпроизводные ряда 2,2,6,6-ТМП. Известно, что N-галогенпроизводные алифатических аминов применяются в органическом синтезе в качестве галогенирующих, дегидрирующих и аминирующих агентов, но их использование ограничено трудностью получения и низкой стабильностью. N-галогенпроизводные ряда 2,2,6,6-ТМП достаточно стабильны, однако методы их синтеза, свойства и области их применения изучены недостаточно. Уникальность этих соединений связана с пространственным экранированием атома азота, что приводит к ряду необычных свойств.
В связи с этим актуальными являются исследования по электрохимическому галогенированию и окислению ПЗА ряда 2,2,6,6-ТМП – предшественников наиболее широко используемых НР, генерированию НР в процессах электрохимического окисления органических веществ из 1-галогенаминов и самих ПЗА ряда 2,2,6,6-ТМП, разработки более простых и экономичных методов синтеза соединений рядов ТМП, тетраметилпирролина и тетраметилпирролидина, а также изучение новых областей практического приложения полученных соединений в органическом синтезе и в каталитических процессах непрямого электрохимического окисления органических соединений.
Применение НР ряда 2,2,6,6-ТМП и 2,2,5,5-тетраметилпирролидина ограничено трудностью их получения и высокой стоимостью. Проведенные исследования расширят возможности производства ПЗА и НР этих рядов, относящихся к продуктам малотоннажной химии, и будут способствовать увеличению их ассортимента. По разработанным методикам организовано получение реактивов по заказам научно-исследовательских лабораторий НИИ в количестве до 1 кг.
Исследования проводились на кафедре химической технологии высокомолекулярных соединений, органической, физической и коллоидной химии Южно-Российского государственного технического университета (Новочеркасского политехнического института) в рамках научного направления университета «Синтез новых соединений с заданными свойствами и источников энергии» по г/б теме № 1.05 «Разработка теоретических основ синтеза новых химических соединений с заданными свойствами и способов их получения», а также в Институте органической и физической химии Казанского научного центра РАН.
Цель работы.
Научное обоснование и разработка эффективных электрохимических методов галогенирования и окисления пространственно-затрудненных аминов, нитроксильных радикалов ряда 2,2,6,6-тетраметилпиперидина и на их основе новых путей синтеза практически важных соединений ряда 2,2,6,6-ТМП и 2,2,5,5-тетраметилпирролидина, изучение их свойств и областей использования.
В соответствии с поставленной целью решались следующие задачи:
- разработка препаративных синтезов 1-галогенаминов ряда 2,2,6,6-ТМП. Изучение стабильности полученных соединений;
- исследование процессов электрохимического окисления ПЗА и 1-галогенаминов ряда 2,2,6,6-ТМП;
- изучение редокс-свойств и стабильности НР ряда 2,2,6,6-ТМП в зависимости от их строения и состава электролита;
- разработка препаративных методов непрямого каталитического электрохимического окисления спиртов с применением НР, аминов и 1-галогенаминов ряда 2,2,6,6-ТМП;
- экспериментальное обоснование возможности применения 1-галогенпроизводных ряда 2,2,6,6-ТМП в качестве окислителей и галогенирующих агентов;
- исследование процессов электрохимического хлорирования и бромирования ТАА, выявление влияния различных факторов на направление электрохимического галогенирования ТАА;
- разработка одностадийных препаративных методов синтеза 3-карбоксамидо-2,2,5,5-тетраметилпирролидина путем электрохимического галогенирования ТАА в щелочной среде и 3-метокси(этокси-)2,2,5,5-тетраметилпирролидинов в процессе электрохимического галогенирования ТАА в растворе метилата(этилата) натрия;
Научная новизна.
• Установлено, что при электрохимическом галогенировании ТАА и других соединений ряда 2,2,6,6-ТМП в нейтральной среде в присутствии органического растворителя образуются 1-галогенпроизводные ряда 2,2,6,6-ТМП;
• Впервые при электрохимическом окислении 2,2,6,6-ТМП получен свободный НР – 2,2,6,6-ТМП-1-оксил. Предложен механизм этого процесса;
• Впервые показано, что процесс электрохимического окисления 1-галогенпроизводных ряда 2,2,6,6-ТМП является обратимым и приводит к образованию соответствующих катион-радикалов, что подтверждено данными ЦВА и ЭПР;
• При электрохимическом окислении ПЗА ряда 2,2,6,6-ТМП методом ЭПР зафиксировано образование соответствующих аминильных радикалов и НР, которые ранее наблюдали только в фотохимических и химических реакциях N-замещенных производных 2,2,6,6-ТМП;
• Впервые показана возможность использования аминов и 1-галогенаминов ряда 2,2,6,6-ТМП в качестве катализаторов в реакциях непрямого электрохимического окисления спиртов, а также возможность использования 1-галогенаминов ряда 2,2,6,6-ТМП в качестве окислителей фенолов и галогенирующих агентов ароматических соединений;
• Установлено, что при использовании каталитической системы НР ряда 2,2,6,6-ТМП – йодид калия электрохимическое окисление спиртов протекает селективно с образованием из первичных спиртов – альдегидов, из вторичных – кетонов, с высоким выходом по веществу и по току;
• Впервые обнаружено, что электрохимическое галогенирование ТАА в кислой среде приводит к образованию -галогензамещенных пиперидонов – важных промежуточных продуктов в синтезе НР ряда 2,2,5,5-тетраметилпирролидина;
• Установлено, что электрохимическое галогенирование ТАА в щелочной среде сопровождается перегруппировкой Фаворского и приводит к образованию с высоким выходом смеси продуктов ряда 2,2,5,5-тетраметилпирролина и 2,2,5,5-тетраметилпирролидина. Эта реакция протекает с количественным превращением ТАА и открывает путь к самому удобному из известных методов получения этих соединений. Разработаны методы восстановления смеси амидов и эфиров ряда пирролина и пирролидина с целью получения чистых 3-карбоксамидо-, 3-карбметокси- или 3-карбэтокси-2,2,5,5-тетраметилпирролидинов.
Практическая значимость.
• Получены новые данные о свойствах 1-галогенпроизводных ряда 2,2,6,6-ТМП. Показано, что эти соединения могут использоваться как эффективные галогенирующие и дегидрирующие агенты ароматических соединений. Проведенные исследования также позволяют использовать 1-галогенамины ряда 2,2,6,6-ТМП и их структурные предшественники – ПЗА в каталитических процессах непрямого электрохимического окисления первичных и вторичных спиртов;
• На основе полученных данных по механизму электрохимического окисления 1-хлор(бром)-2,2,6,6-ТМП разработан препаративный метод электрохимического получения 2,2,6,6-ТМП-1-оксила с высоким выходом по току и веществу (80-85 % на вступивший в реакцию 1-хлор(бром)-2,2,6,6-ТМП);
• Разработан препаративный метод непрямого каталитического электрохимического окисления спиртов каталитической системой НР – йодид калия, позволяющий проводить в мягких условиях селективное окисление спиртов с высоким выходом по току и веществу;
• Разработаны методы синтеза 3,5-дибром-4-оксо-2,2,6,6-ТМП, 3-карбоксамидо-2,2,5,5-тетраметилпирролидина и на их основе 3,5-дибром-4-оксо-2,2,6,6-ТМП-1-оксила и 3-карбоксамидо-2,2,5,5-тетраметилпирролидин-1-оксила; 3-карбметокси-2,2,5,5-тетраметилпирролидина; 3-карбэтокси-2,2,5,5-тетраметил-пирролидина. Получены моно- и бирадикалы, которые могут быть использованы в качестве добавки к активной массе в источниках тока. Разработаны методики анализа, с помощью которых можно определять количественный состав смеси продуктов. На основе полученных в диссертации данных разработана научно-техническая документация на производство реактивов 7 наименований (лабораторные методики и технические условия);
• Результаты проведенных исследований применяются при выполнении дипломных работ, НИР студентов и в лабораторном практикуме по курсам «Органический синтез», «Основы физико-химии полимеров», «Органическая химия», «Основные методы научных исследований».
Личный вклад автора.
Постановка темы и задач исследований, обработка, анализ полученных результатов принадлежат автору. Все результаты, приведенные в диссертации, получены либо самим автором, либо под его руководством, либо при его непосредственном участии. ЭПР-спектры катион-радикалов получены в институте органической и физической химии Казанского научного центра РАН.
Основные положения, вынесенные на защиту.
• Электрохимические методы синтеза 1-галогенпроизводных ряда 2,2,6,6-ТМП.
• Механизм процессов окисления ПЗА и 1-галогенаминов ряда 2,2,6,6-ТМП;
• Электрохимический метод синтеза 2,2,6,6-ТМП-1-оксила; исследование механизма процесса образования этого НР;
• Использование 1-галогенаминов ряда 2,2,6,6-ТМП в качестве окислителей, галогенирующих и дегидрирующих реагентов в органическом синтезе;
• Редокс-свойства НР ряда 2,2,6,6-ТМП в зависимости от их строения и состава электролита;
• Непрямое электрохимическое окисление спиртов в присутствии ПЗА, 1-галогенаминов и НР ряда 2,2,6,6-ТМП;
• Электрохимические методы синтеза 3,3,5,5-тетрахлор-4-оксо-2,2,6,6-ТМП; 3,5-дибром-4-оксо-2,2,6,6-ТМП; методы синтеза аминов и НР на их основе;
• Электрохимический синтез 3-карбоксамидо-, 3-карбметокси-, 3-карбэтокси-2,2,5,5-тетраметилпирролидинов;
Апробация работы.
Материалы диссертационной работы доложены и обсуждены на: III Всесоюзном совещании по химическим реактивам, г.Ашхабад, 1989; XII Всесоюзном совещании по электрохимии органических соединений, г. Караганда, 1990; Семинаре-совещании «Потребители и производители органических реактивов», г. Дилижан, Армения, 1991; IV Всесоюзном совещании по химическим реактивам, г. Баку, 1991; XIII Всесоюзном совещании по электрохимии органических соединений, г. Тамбов, 1994; 9-ой Европейской конференции по электроорганической химии, г. Сан-Фелио, Испания, 1995; VI Всесоюзной конференции «Карбонильные соединения в синтезе гетероциклов», Саратов, 1996; Европейской исследовательской конференции «Органическая электрохимия», г. Агелонде, Франция, 1998; Международной конференции «Органическая электрохимия в новом веке», г. Томар, Португалия, 2000; Международной конференции «Актуальные проблемы органической химии», г. Новосибирск, 2001; Всероссийской научно-практической конференции «Химия в технологии и медицине», г. Махачкала, 2002; Всероссийской научно-практической конференции «Электрохимия органических соединений», г. Астрахань, 2002; Международной научной конференции «Химия, химическая технология и биотехнология на рубеже тысячелетий», г. Томск, 2006; Всероссийской конференции «Электрохимия и экология», г. Новочеркасск, 2008 г; IX Международном семинаре по магнитному резонансу: спектроскопия, томография и экология, г. Ростов-на-Дону, 2008; I Международной конференции «Новые направления в химии гетероциклических соединений» г. Кисловодск, 2009.
Публикации. Результаты проведенных исследований представлены в 56 печатных работах, в том числе в 18 статьях, опубликованных в журналах, рекомендованных ВАК, в 2 патентах РФ и 3 монографиях.
Структура и объем диссертации. Диссертационная работа состоит из 10 глав, введения, выводов и списка цитируемой литературы, состоящего из 365 наименований; изложена на 332 страницах машинописного текста без приложений, включает 75 схем, 19 таблиц и 70 рисунков.
Основное содержание работы
Во введении излагается актуальность темы исследований, сформулированы цель и задачи диссертации.
В главе 1 (обзор литературных данных) изложены данные по галогенированию соединений ряда 2,2,6,6-ТМП, в том числе ТАА. Далее рассмотрены процессы окисления алифатических аминов, 1-галогенаминов и НР ряда 2,2,6,6-ТМП. Особое внимание уделено способам генерирования и механизмам образования неароматических аминильных радикалов и катион-радикалов аминов. Подробно проанализированы работы, посвященные электрохимии НР ряда 2,2,6,6-ТМП и их использованию в качестве катализаторов (медиаторов) в процессах окисления органических соединений.
В главе 2 рассмотрены объекты, оборудование и приборы, используемые для исследований, описаны методы подготовки электродов и приготовления растворов. Для решения поставленных задач применяли следующие физико-химические методы: циклическая вольтамперометрия (ЦВА); потенциометрия; ЭПР- УФ-; ИК-; ЯМР-спектроскопия; элементный анализ; хроматографические методы – тонкослойная хроматография (ТСХ), газо-жидкостная хроматография (ГЖХ); препаративный электролиз.
ЦВА-кривые получали с помощью потенциостата ПИ-50 с программатором ПР-8; потенциалы, приведенные в работе, измерены в водной среде отн. Ag/AgCl – электрода, в ацетонитриле отн. Ag/0,01 M AgNO3 - электрода. ЭПР-спектры регистрировали с помощью радиоспектрометра “Radiopan SE/X-2544”; (расчет ЭПР-спектров проведен с использованием программы «WinSIM EPR DESIGN V.9,5»), УФ-спектры снимали на приборе "Specord UV VIS", ИК-спектры – на приборе «Specord 75 IR» (в вазелиновом масле или жидкой пленке), ЯМР-спектры – на приборе "Varian VXR-300", продукты электролиза анализировали с помощью газового хроматографа “ЛХМ-8 МД”.
В главах 3-9 изложено основное содержание работы. В главе 10 приведены разработанные автором методики синтезов и методики количественного анализа смеси пиперидинов, пирролинов и пирролидинов.
Глава 3. Электрохимический синтез и свойства 1-галогензамещенных соединений ряда 2,2,6,6-тетраметилпиперидина (2,2,6,6-ТМП)
3.1 Электрохимическое галогенирование соединений ряда 2,2,6,6-ТМП
Электрохимический синтез 1-галогенпроизводных ряда 2,2,6,6-ТМП (5-12) (схема 1) из пространственно-затрудненных аминов (ПЗА) (1-4) проводили как в диафрагменном, так и в бездиафрагменном электролизере с использованием Pt-электродов при плотности тока 0,1 – 0,15 А/см2 (при хлорировании) и 0,05 – 0,1 А/см2 (при бромировании), в двухфазной системе хлористый метилен – водный раствор NaCl или NaBr, количество пропущенного электричества 2 F. 1-Галогенамины (5-12) образуются с высоким выходом по веществу (80-95 %) и току (75-90 %). Выход целевых продуктов в диафрагменном электролизере на 10-15 % выше, чем в электролизере без диафрагмы. Строение полученных веществ подтверждено данными элементного анализа и ПМР-спектроскопии.

Схема 1 – Электрохимическое галогенирование производных ряда 2,2,6,6-ТМП
Единственным продуктом реакции электрохимического галогенирования 2,2,6,6-ТМП (1) в бездиафрагменном электролизере в интервале рН 6-7 после пропускания теоретического количества электричества является 1-хлор- (5) или 1-бром-2,2,6,6-ТМП (9). Однако, после пропускания дополнительного количества электричества (4 F) образуется 2,2,6,6-ТМП-1-оксил (13) и оксоаммониевая соль (14) (схема 2). Был предложен механизм реакции образования НР (13) (схема 5, с. 11), основанный на данных ЦВА и ЭПР-спектроскопии, анализе полученных продуктов и литературных данных.

Схема 2 – Электрохимическое превращение 2,2,6,6-ТМП (1) в НР (13)
Электрохимическое бромирование 1,2,5,6-тетрагидро-2,2,6,6-тетраметилпиридина (15) в слабокислой среде (рН 3-5) приводит к образованию 3,4-ди-бром-2,2,6,6-ТМП (17) с выходом 80 % (схема 3). По-видимому, образующийся вначале реакции 1-бром-1,2,5,6-тетрагидро-2,2,6,6-тетрагидропиридин (16) бромирует исходное соединение с образованием 3,4-дибром-2,2,6,6-ТМП (17). Строение полученного препарата подтверждено встречным синтезом. Эта реакция протекает только в слабокислой среде. В нейтральной среде образуется гидробромид 1,2,5,6-тетрагидро-2,2,6,6-тетраметилпиридина. В кислой среде (рН 1-2) реакция не проходит.

Схема 3 - Электрохимическое бромирование 1,2,5,6-тетрагидро-2,2,6,6-тетраметилпиридина (15)
Электрохимическое галогенирование 4-гидроксимино-2,2,6,6-ТМП (18) приводит к образованию 4-нитрозо-4-хлор-2,2,6,6-ТМП гидрохлорида (19), который далее окисляется кислородом воздуха в 4-нитро-4-хлор-2,2,6,6-ТМП гидрохлорид (20). Свободное основание (21) может быть получено при действии на соединение (20) 10 %-ного раствора карбоната натрия (схема 4).

Схема 4 - Электрохимическое хлорирование 4-гидроксимино-2,2,6,6-ТМП (18)
* - Окисление можно провести кислородом воздуха или перекисью водорода в щелочной среде
Реакцию проводили в диафрагменном электролизере в кислой среде (рН 3-4) на Pt-аноде при плотности тока 0,05-0,10 А/см2 в присутствии хлористого метилена. 4-Нитрозо-4-хлор-2,2,6,6-тетраметилпиперидин гидрохлорид (19) был получен с выходом по веществу 70 % после пропускания 4 F электричества. Вероятно, в реакции хлорирования 4-гидроксимино-2,2,6,6-ТМП (18) хлорирующим агентом является 1-хлор-4-гидроксимино-2,2,6,6-ТМП (22), который образуется при хлорировании соединения (18) молекулярным хлором. Образование промежуточного соединения (22) подтверждается сравнением ЦВА процесса окисления модельного соединения 1-хлор-2,2,6,6-ТМП (5) (рисунок 1) и процесса хлорирования соединения (18) (рисунок 2).
На рисунке 1 приведена ЦВА 1-хлор-2,2,6,6-ТМП (5) в 1,5 М HClO4, где при потенциале 2,0 В наблюдается максимум, связанный с окислением 1-хлор-2,2,6,6-ТМП (5) на Pt. На рисунке 2 на ЦВА-кривой - два максимума: один при потенциале 1,4 В, другой – 2,0 В. Первый обусловлен разрядом Cl-, второй, по-видимому, окислением 1-хлор-4-гидроксимино-2,2,6,6-ТМП (22), который образуется в результате взаимодействия хлора и соединения (18), введенного в электролит. ЦВА на рисунках 1 и 2, начиная с потенциала 1,6 В полностью совпадают, что подтверждает образование соединения (22) в качестве промежуточного в синтезе 4-нитрозо-4-хлор-2,2,6,6-ТМП гидрохлорида (19).
Реакция бромирования соединения (18) протекает при рН 5-7 аналогично хлорированию, в бездиафрагменном электролизере и приводит к образованию 4-бром-4-нитро-2,2,6,6-ТМП гидробромида. Строение полученного гидробромида подтверждено данными элементного анализа и ПМР-спектроскопии.
Рисунок 1 - ЦВА 1. 10-3 М 1-хлор-2,2,6,6-ТМП (5) в среде 1,5 М HСlO4 на Pt-аноде; = 0,1 В/с
Рисунок 2 - ЦВА 210-4 М 4-гидроксимино-2,2,6,6-ТМП (18) в среде 1,5 М HСlO4 и 1. 10-3 М NaCl на Pt-аноде; = 0,1 В/с
3.2 Изучение стабильности 1-галогенаминов ряда 2,2,6,6-ТМП
Полученные в процессе электрохимического галогенирования ПЗА соединения (3, 5, 8, 9, 11, 12) (схема 1, с. 8) являются достаточно устойчивыми в процессе хранения, за исключением соединений (6, 10), которые при хранении через 2-3 суток постепенно превращаются в хлор- или бромгидраты ТАА (2) соответственно. Особо устойчивыми являются соединения (5, 9), которые, в определенных условиях хранения (отсутствие влаги, температура 5-10 0С) сохраняют свою структуру длительное время. Однако количественные данные о стабильности этих соединений отсутствуют. Цель данной части работы заключалась в изучении стабильности галогенаминов на примере 1-хлор-2,2,6,6-ТМП (5) и 1-хлор-4-гидрокси-2,2,6,6-ТМП (7) в зависимости от продолжительности хранения в метаноле и этаноле, изменения pH среды и действия УФ-излучения.
Для определения концентрации соединений (5) и (7) в исследуемых растворах были использованы спектральный метод анализа (УФ-спектроскопия) и иодометрический метод определения свободного хлора. Спектры регистрировались при длине волны (max) 285 нм для соединения (5) и 270 нм для соединения (7). Расхождение между данными, полученными спектрофотометрическим методом анализа и йодометрическим титрованием не превышает 5 %. Облучение раствора соединения (7) УФ-светом приводит к увеличению скорости распада этого соединения. Меньшая стабильность (7) в метанольных растворах при хранении в темноте и облучении, возможно, связана с образованием внутримолекулярной водородной связи. Образование этой связи изменяет конформацию молекулы и ее реакционную способность.
При добавлении растворов соединений (5) или (7) в метаноле к буферным растворам с различным значением pH обнаружено, что изменение рН раствора приводит к изменению скорости превращения 1-хлор-2,2,6,6-тетраметилпиперидина (5) и 1-хлор-4-гидрокси-2,2,6,6-ТМП (7). При увеличении кислотности среды скорость распада соединений (5) и (7) увеличивается. Следует отметить, что в водно-метанольных растворах стабильность соединений (5) и (7) практически не отличается, так как в этих условиях внутримолекулярная водородная связь не реализуется.
Глава 4. Электрохимическое окисление 1-галогенаминов и аминов ряда 2,2,6,6-тетраметилпиперидина (2,2,6,6-ТМП)
4.1 Электрохимическое окисление 1-галогенаминов ряда 2,2,6,6-ТМП
В этой части работы методами ЦВА, ЭПР и препаративного электролиза изучена реакция электрохимического окисления 1-хлор-2,2,6,6-ТМП и других пространственно-затрудненных аминов (ПЗА) ряда 2,2,6,6-ТМП. Единственным продуктом реакции электрохимического галогенирования 2,2,6,6-ТМП (1) в бездиафрагменном электролизере в интервале рН 6-7 после пропускания теоретического количества электричества (2 F) является 1-хлор- (5) или 1-бром-2,2,6,6-ТМП (9). Установлено, что пропускание дополнительного количества электричества (4 F) приводит к образованию 2,2,6,6-ТМП-1-оксила (13) (глава 3, раздел 3.1, схема 2, с. 8).
Это случайное наблюдение явилось основанием для дальнейшего исследования этой реакции. Был разработан препаративный метод синтеза нитроксильного радикала (НР), основанный на электрохимическом окислении гидрохлорида 2,2,6,6-ТМП (1) или 1-галогенаминов (5, 9) в двухфазной системе CH2Cl2-H2O. Электрохимический синтез НР (13) можно проводить как в бездиафрагменном электролизере, так и в диафрагменном, используя в качестве исходного амин (1). В последнем случае следует использовать фосфатный буфер (рН 6,8), так как в отсутствии буфера происходит закисление электролита. При проведении электролиза в органической фазе остается не вступивший в реакцию 1-галогенамин (5), а в водной фазе находится оксоаммониевая соль (14). Для выделения продукта реакции – НР (13) оксоаммониевую соль (14) восстанавливали нитритом натрия, гидроксидом натрия или электрохимически. НР (13) хорошо растворяется в органических растворителях и может быть легко выделен из водной фазы. Это первый и пока единственный пример электрохимического окисления ПЗА ряда 2,2,6,6-ТМП, приводящий к образованию НР (13). Выход 2,2,6,6-ТМП-1-оксила (13) составляет 82 % по веществу (на вступивший в реакцию 1-хлор-2,2,6,6-ТМП (5)). Необходимым условием реализации данного способа синтеза НР является наличие в растворе в качестве исходного соединения или интермедиата галогенаминов типа (5). Их образование представляет изученный процесс галогенирования ПЗА (глава 3), который осуществляется достаточно просто. В этих же условиях, но в отсутствии галогенид-ионов в электролите, окисление ПЗА ряда 2,2,6,6-ТМП не происходит. В результате препаративного электролиза были выделены только исходные амины.
Процесс образования НР (13) при электрохимическом окислении 1-хлор- (5) или 1-бром-2,2,6,6-ТМП (9) был неясен. Основываясь на данных ЦВА и ЭПР предложен механизм процесса образования НР (13) и иона оксоаммония (14) при окислении 1-хлорамина (5) (схема 5):

Схема 5 – Механизм окисления 1-хлор-2,2,6,6-ТМП (5) до НР (13) и оксоаммониевого катиона (14)
Механизм включает стадии образования катион-радикала (23), элиминирование молекулы хлора с образованием катиона (24), с последующим гидроксилированием и образованием соединения (25), элиминирование протона и электронный перенос.
Анализ ЦВА-кривых 1-хлор-2,2,6,6-ТМП (5) (рисунок 3) показал, что процесс окисления является обратимым, одноэлектронным и приводит к образованию соответствующего катион-радикала (23). Это подтверждается линейной зависимостью ip от 1/2 (рисунок 4) и величиной Ер=60 мВ, характерной для обратимого одноэлектронного процесса. Катион-радикал (23) (схема 5) во временной шкале записи ЦВА-кривых (секунды) вступает в химические реакции, на что указывает снижение относительной высоты пика ре-восстановления с уменьшением скорости развертки потенциала. Введение воды (10 %) в электролит практически не отражается на обратимости пика окисления, что свидетельствует о медленной скорости взаимодействия катион-радикала (23) с водой.
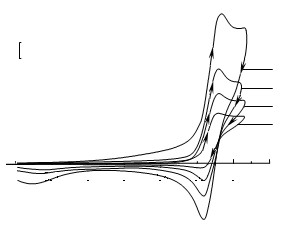
Рисунок 3 – ЦВА 3·10-3 M 1-хлор-2,2,6,6-ТМП (5) на СУ-аноде в среде CH3CN/0,1 M Bu4NBF4 при разных скоростях развертки потенциала: 1 – 0,2 В/с; 2 – 0,1 В/с; 3 – 0,05 В/с; 4 – 0,02 В/с
Рисунок 4 – Зависимость тока анодного пика 3·10-3 M 1-хлор-2,2,6,6-ТМП (5) от корня квадратного из скорости развертки потенциала в среде CH3CN/0,1 M Bu4NBF4
На обратной ветви ЦВА в анодной области потенциалов кроме пика восстановления катион-радикала (23) исходного галогенамина (5) в области 0,1-0,2 В регистрируется второй пик, относящийся к восстановлению иона оксоаммония (14) до НР (13). В случае бромамина (9) удалось зафиксировать и небольшой пик окисления НР (13) при повторной записи ЦВА сразу же после ре-восстановления. Таким образом, данные ЦВА свидетельствуют о том, что при одноэлектронном электрохимическом окислении галогенаминов (5) и (9) образуются катион-радикалы, претерпевающие последующие превращения с образованием в качестве одного из продуктов соответствующих ионов оксоаммония (14), восстановление которых приводит к НР (13).
Данные ЦВА подтверждаются результатами ЭПР-спектроскопии. При электроокислении 1-хлор-2,2,6,6-ТМП (5) в ацетонитриле на Pt-электроде непосредственно в резонаторе ЭПР-спектрометра при пониженной температуре (243 К) удалось зафиксировать спектр катион-радикала (23) (рисунок 5), состоящий из 12 линий со следующими характеристиками: g = 2,0093, aN = 20,9 э – константа сверхтонкого взаимодействия (СТВ) неспаренного электрона с ядром 14N, a35Cl = 5,71 э, a37Cl = 4,75 э – константы СТВ с ядрами изотопов хлора. На рисунке 6 представлен ЭПР-спектр катион-радикала соединения (9). Катион-радикал (23) достаточно устойчив (продолжительность жизни при 243 К составляет ~ 10 мин.). Стабильность катион-радикала бромамина (9) ниже (время жизни ~ 3 мин.). При обратном восстановлении продуктов окисления при 0 В зафиксирован интенсивный сигнал НР (13). Какие-либо иные радикальные частицы в этих условиях методом ЭПР не обнаружены.
Рисунок 5 – ЭПР-спектр катион-радикала 1-хлор-2,2,6,6-ТМП (5): 1 – спектр катион-радикала (23), полученный экспериментальным путем; 2 – спектр катион-радикала (23), рассчитанный на основании следующих данных: g = 2,0093, aN = 20,9 э – константа СТВ неспаренного электрона с ядром 14N, a35Cl = 5,71 э и a37Cl = 4,75 э – константы СТВ неспаренного электрона с ядрами изотопов хлора
Рисунок 6 - ЭПР-спектр катион-радикала 1-бром-2,2,6,6-ТМП (9): 1 – спектр катион-радикала, полученный экспериментальным путем; 2 – спектр катион-радикала, рассчитанный на основании следующих данных: g = 2,023, aN = 21 э – константа СТВ неспаренного электрона с ядром 14N, a79Br = 18,5 э и a81Br = 19,9 э – константы СТВ с ядрами изотопов брома
Аналогичные результаты были получены при изучении процесса электрохимического окисления галогенаминов (7, 8, 11, 12). ЦВА этих галогенаминов, полученные в среде MeCN/0,1 M Bu4NBF4 на СУ-электроде, в целом подобны ЦВА кривым галогенаминов (5, 9). Для всех изученных галогенаминов (5, 7-9, 11, 12) независимо от природы галогена ток пика окисления закономерно снижается с уменьшением скорости развертки потенциала в интервале от 0,20 до 0,02 В/с и зависимость ip от 1/2 линейна, что свидетельствует о диффузионной природе тока. На обратной ветви ЦВА при всех скоростях развертки потенциала регистрируется интенсивный пик ре-восстановления, сдвинутый на 0,06-0,07 В относительно пика окисления в сторону менее положительных потенциалов, что указывает на обратимость как самой стадии электронного переноса, так и электродного процесса одноэлектронного окисления в целом, т.е. на достаточную стабильность образующихся катион-радикалов галогенаминов (5, 7-9, 11, 12) за время вольтамперных измерений (секунды). Катион-радикалы галогенаминов (7, 8, 11, 12) непосредственно зарегистрировали методом ЭПР-спектроскопии, проводя их электроокисление в ячейке в резонаторе ЭПР-спектрометра на Pt-электроде при потенциале +1,1 В для бромаминов (11, 12) и +1,3 В для хлораминов (7, 8) при пониженной температуре (263 К). После завершения процесса окисления и при последующем ре-восстановлении (при 0 В) методом ЭПР регистрируется интенсивный сигнал, появляющегося в растворе соответствующего НР. Какие-либо иные радикальные частицы методом ЭПР не обнаруживаются.
4.2 Электрохимическое окисление пространственно-затрудненных аминов (ПЗА) ряда 2,2,6,6-тетраметилпиперидина (2,2,6,6-ТМП)
Одним из важных следствий изучения свойств НР, впервые полученных в 1960-х годах, явилось открытие нового класса светостабилизаторов полимеров – соединений ряда 2,2,6,6-ТМП. Синтезу, изучению свойств и применению светостабилизаторов этого ряда посвящено огромное количество исследований. Однако механизм их действия до сих пор остается спорным. Полученные ранее данные не дают ответа на вопрос, являются ли ПЗА ряда 2,2,6,6-ТМП истинными стабилизаторами, или они только генерируют частицы, которые прерывают окислительную деструкцию полимеров. Поэтому изучение механизма окисления ПЗА представляет теоретический и практический интерес. Кроме этого, образующиеся при окислении ПЗА промежуточные продукты могут быть использованы в качестве медиаторов при проведении реакций окисления органических соединений, а также для синтеза НР.
В этой части работы изучен механизм электрохимического окисления аминов ряда 2,2,6,6-ТМП. В процессе электрохимического окисления промежуточные продукты были идентифицированы методами ЦВА и ЭПР-спектроскопии. В качестве модельных соединений были использованы амины (1-4). Проведенные в работе исследования аминов ряда 2,2,6,6-ТМП показали, что электрохимическое поведение ПЗА (1-4) отличается от поведения 1-галогенпроизводных этого же ряда.
На ЦВА-кривых изученных пиперидинов (1-4), полученных в ацетонитриле на фоне 0,1 M Bu4NBF4 в инертной атмосфере (аргон) на СУ-электроде, в анодной области наблюдается по два необратимых пика окисления. Первый пик в интервале потенциалов от +0,72 до +0,90 В (таблица 1). Он соответствует потенциалу окисления вторичных аминов до катион-радикалов согласно литературным данным. Второй пик сдвинут относительно первого на 0,18 – 0,40 В.
Таблица 1 – Потенциалы* пиков окисления ПЗА (1-4) в среде MeCN/0,1 M Bu4NBF4 на СУ-электроде (С = 210-3 М, = 0,1 В/с, Т = 295 К)
| Соединение | (1) | (2) | (3) | (4) |
| Ep,ox, В | +0,90; +1,18** | +0,72; +1,14** | +0,73; +0,91** | +0,80; +0,98** |
* потенциалы измерены и приведены относительно Ag/0,01 M AgNO3 в MeCN;
** в присутствии 10 % Н2О
Группы, вводимые в 4-е положение пиперидинового кольца, влияют на потенциалы окисления в соответствии с их электронными свойствами: акцепторные заместители затрудняют, а донорные – облегчают окисление аминов. При введении в раствор молекулярного кислорода до насыщения, воды до 10 %, морфология ЦВА-кривых практически не меняется.
Более детально электрохимическая реакция окисления ПЗА была исследована на примере 4-оксо-2,2,6,6-ТМП (2). Прежде всего, зафиксированы ЦВА с реверсом потенциала с обоих пиков окисления (рисунки 7 а, б). При реверсе с потенциала первого пика окисления (рисунок 7 а) на обратной ветви ЦВА-кривой фиксируется обратимый пик ре-восстановления, по потенциалу (0,4 В) соответствующий восстановлению катиона оксоаммония в редокс-паре 4-оксо-2,2,6,6-ТМП-1-оксил (26) - ион оксоаммония (27) (схема 6, с. 16).
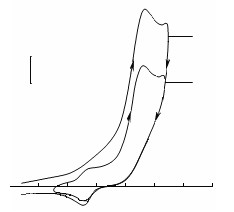

а) б)
Рисунок 7 – ЦВА 310-3 M 4-оксо-2,2,6,6-ТМП (2) на СУ-аноде в среде CH3CN/0,1 M Bu4NBF4, = 0,1 В/с: а) в интервале потенциалов от 0 В до 1,2 В: 1 – первый анодный цикл; 2 – второй анодный цикл; б) в интервале потенциалов от 0 В до 1,5 В: 1 – первый анодный цикл; 2 – второй анодный цикл
Следовательно, при потенциалах первого пика происходит окисление ТАА (2), одним из продуктов которого является соответствующий ион оксоаммония (27). Если обратную развертку начинать с потенциалов второго пика окисления (рисунок 7 б) интенсивность пика восстановления иона оксоаммония (27) снижается. Кроме того, при скорости развертки потенциала 0,20 В/с и расширении интервала сканирования в пределах от -1,2 до +1,4 В удается зафиксировать слабые пики ре-восстановления при -0,65 и -0,81 В (рисунок 8), первый из которых отнесли к восстановлению аминильного радикала (28), а второй – к восстановлению НР (26). Введение воды (10 %) приводит к увеличению интенсивности пика восстановления иона оксоаммония (27), причем интенсивность сохраняется постоянной при реверсе потенциала с первого и со второго пиков окисления.
ЦВА соединения (2) на Pt-электроде аналогичны кривым на СУ, что свидетельствует об идентичности превращений на обоих электродах и независимости процесса окисления от материала электрода.
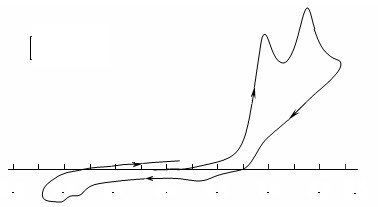
Рисунок 8 – ЦВА 1·10-2 M 4-оксо-2,2,6,6-ТМП (2) на СУ-аноде в среде CH3CN/0,1 M Bu4NBF4 в интервале потенциалов от -1,0 В до 1,4 В; = 0, 2 В/с
Исходя из данных ЦВА окисление амина (2) до НР (26) и далее до катиона оксоаммония (27) в ацетонитриле может протекать по ЕСЕС-механизму (схема 6).

Схема 6 – Механизм окисления 4-оксо-2,2,6,6-ТМП (2)

Электроокисление амина (2) на Pt-электроде непосредственно в резонаторе ЭПР-спектрометра в ацетонитриле на фоне 0,1 M Bu4NBF4 при потенциалах первого пика окисления при комнатной и пониженной температуре (243 К) в начальный момент времени приводит к синхронному быстрому росту интенсивности ЭПР-сигналов соответствующих аминильного (28) (g = 2,0043 aN = 14,9 э, a12H = 1,12 э, a4C13 = 13 э) и нитроксильного (26) (g = 2,0059, aN = 14,7 э, aH < 0,06 э, a4C13 = 5,4 э) радикалов (рисунок 9). Далее в процессе электролиза соотношение между концентрациями радикалов меняется. Концентрация НР (26) после достижения определенного предельного значения дальше не растет, а концентрация аминильного радикала (28) продолжает расти и через некоторое время (~10 мин.) ЭПР фиксирует практически только один аминильный радикал (28) (рисунок 10).
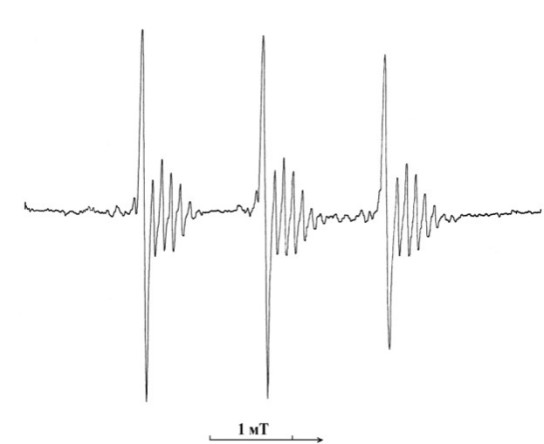
Рисунок 9 – ЭПР-спектр НР (26) и аминильного радикалов (28), генерируемых электроокислением 4-оксо-2,2,6,6-ТМП (2) (С = 310-3 М) на Pt-электроде непосредственно в резонаторе ЭПР-спектрометра в электролите MeCN/ 0,1M Bu4NBF4 при потенциалах первого пика окисления; (Т=243 К)
Рисунок 10 – ЭПР-спектр аминильного радикала (28), генерируемого электроокислением 4-оксо-2,2,6,6-ТМП (2) (С = 2,510-3 М) на Pt-электроде непосредственно в резонаторе ЭПР-спектрометра в среде MeCN/0,1 M Bu4NBF4 в инертной атмосфере аргона при потенциалах первого пика окисления через 10 мин. после начала электролиза; (Т=243 К), края спектра прописаны с усилением в 10 раз
После отключения электрического тока аминильный радикал (28) достаточно быстро погибает и фиксируется только устойчивый НР (26), интенсивность сигнала которого усиливается во времени (рисунок 11). В присутствии воды и кислорода в целом наблюдается аналогичная картина. Отличие заключается только в том, что введение 10 % воды в ацетонитрил приводит к увеличению доли НР (26) и уменьшению интенсивности сигнала аминильного радикала (28). Образовавшийся в начале электролиза НР (26) окисляется до катиона оксоаммония (27) и восстанавливается на катоде до соответствующего гидроксиламина (31) (схема 6). При проведении препаративного электролиза при потенциале 1,2 В в ацетонитриле или в двухфазной системе CH2Cl2 – водный раствор Na2SO4 после пропускания 2 F электричества из электролита удается выделить только исходные ПЗА и некоторое количество смолообразных продуктов. Это подтверждает тот факт, что основной причиной гибели катион-радикалов аминов (1-4) является не превращение их в НР, а другие процессы, например, взаимодействие с ацетонитрилом, восстановление на катоде или взаимодействие соответствующего катион-радикала с аминильным радикалом.

Рисунок 11 – ЭПР-спектр НР (26), полученного в результате электроокисления 4-оксо-2,2,6,6-ТМП (2) (С = 2,510-3 М) на Pt-электроде в резонаторе ЭПР-спектрометра в среде MeCN/0,1 M Bu4NBF4 в атмосфере аргона при потенциалах первого пика окисления и последующей выдержки раствора без электролиза в течение 15 мин; (Т=243 К)
Глава 5. Исследование свойств нитроксильных радикалов (НР) ряда 2,2,6,6-тетраметилпиперидина (2,2,6,6-ТМП) методом ЦВА
Несмотря на большое количество работ по изучению окислительно-восстановительных свойств НР, которые находят применение в качестве реагентов и катализаторов для окисления органических соединений, имеются противоречивые сведения по редокс-потенциалам нитроксильной группы для одних и тех же соединений, приведенные в разных работах. Поэтому данная часть работы выполнена для оценки окислительно-восстановительных потенциалов НР и продуктов их превращений, т.к. от этого зависит эффективность использования НР в реакциях непрямого электрохимического окисления спиртов. Кроме этого данные по окислительно-восстановительным свойствам НР были необходимы для интерпретации механизма процесса образования НР при окислении 1-галогенаминов и аминов ряда 2,2,6,6-ТМП (главы 3 и 4).
В качестве объектов исследования были выбраны наиболее часто используемые НР ряда 2,2,6,6-ТМП (13, 26, 32-35) (схема 7). Для этих НР методом ЦВА определены окислительно-восстановительные потенциалы в водных и неводных средах.

Схема 7 – НР ряда 2,2,6,6-ТМП, использованные для исследований
На ЦВА-кривых НР (13, 26, 32-35), полученных на СУ-электроде в среде MeCN/ 0,1 M Bu4NBF4, фиксируется по одному одноэлектронному пику окисления и восстановления (таблица 2). Пики окисления обратимы и их параметры и обратимость сохраняются в присутствии кислорода, воды (10 %), уксусной кислоты (0,5 М). Следовательно, одноэлектронное окисление НР приводит к образованию оксоаммониевых катионов, стабильных в исследованных средах во временной шкале записи ЦВА-кривых (секунды). Заместители в 4-м положении пиперидинового кольца влияют на потенциал окисления НР в соответствии с их электронными свойствами: донорные снижают потенциал окисления, а акцепторные заместители действуют в противоположном направлении. Пики восстановления НР до анионов гидроксиламинов пологие и необратимые наблюдаются в области потенциалов от -1,65 В до -1,81 В. При добавлении уксусной кислоты (0,5 М) процесс восстановления протекает значительно легче и происходит в области потенциалов от -0,78 до -1,21 В. Слабые пики ре-окисления анионов гидроксиламинов до НР в отсутствии сильного донора протонов фиксируются при отрицательных потенциалах (от -1,27 В до -0,86 В) и явно выраженные пики ре-окисления самих гидроксиламинов при добавлении уксусной кислоты (0,5 М) – в области положительных потенциалов (от +0,86 В до +0,90 В) (таблица 2). Характеристики пиков ре-окисления гидроксиламинов до НР полностью соответствуют замедленному электрохимически необратимому процессу. Пики ре-восстановления НР до анионов гидроксиламинов необратимые, также соответствуют электрохимически необратимому, а химически обратимому переносу электрона на нитроксильную группу. При введении в ацетонитрильный раствор 10 % воды потенциал восстановления НР снижается на 0,2 – 0,4 В, т.е. процесс восстановления происходит существенно легче (таблица 2).
Таблица 2 – Потенциалыa пиков окисления и восстановления и сопряженных с ними пиков ре-восстановления и ре-окисления НР в среде MeCN/ 0,1M Bu4NBF4 на СУ-электроде (С = 210-3 М, = 0,1 В/с, Т = 295 К)
| Соединение | Ep,red, В | Ep,reox, В | Ep,ox, В | Ep,rered, В |
| 13 | -1,79 -1,55б -0,98в - | -0,86 -0,25б +0,95в - | +0,28 - - +0,60г | +0,21 - - +0,55г |
| 26 | -1,75 -1,36б -0,78в | -1,11 -0,83б +0,88в | +0,45 - - | +0,40 - - |
| 32 | -1,72 | -1,08 | +0,31 | +0,24 |
| 33 | -1,65 -1,51б -1,01в - | -1,10 -0,61б +0,90в - | +0,42 - - +0,65г | +0,35 - - +0,58г |
| 34 | -1,81 | -1,27 | +0,30 | +0,22 |
| 35 | -1,75 -1,48б - | -1,22 -0,71б - | +0,39 - +0,62г | +0,32 - +0,56г |
а потенциалы измерены и приведены относительно Ag/0,01 M AgNO3 в MeCN; б в присутствии 10 % Н2О; в в присутствии 0,5 М CH3COOH; г потенциалы измерены в водном растворе электролита (0,05 М Na2SO4) и приведены относительно Ag/AgCl - электрода
На рисунках 12 и 13 приведены ЦВА для 4-оксо-2,2,6,6-ТМП-1-оксила (26) на СУ-электроде в электролите СН3СN/0,1 M Bu4NBF4 в отсутствии доноров протонов (рисунок 12) и c добавкой 0,5 М уксусной кислоты (рисунок 13). На ЦВА-кривых наблюдается обратимый пик для редокс-системы НР (26)/катион оксоаммония (27) (схема 6, с. 16) при потенциалах от +0,45 В до +0,40 В. Пик ре-окисления гидроксиламина (31) (Ep,reox= +0,88 В) свидетельствует о необратимости процесса, и фиксируется после пика окисления НР (26) (рисунок 13).
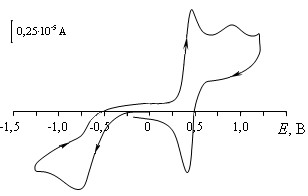
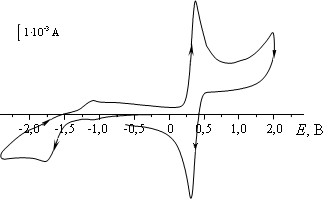
Рисунок 12 – ЦВА 3·10-3 M 4-оксо-2,2,6,6-ТМП-1-оксила (26) на СУ-аноде в среде СН3СN/ 0,1 M Bu4NBF4; = 0,1 В/с
Рисунок 13 – ЦВА 3·10-3 M 4-оксо-2,2,6,6-ТМП-1-оксила (26) на СУ-аноде в среде СН3СN/ 0,1 M Bu4NBF4 и 7,5 10 -4 моль CH3COOH; = 0,1 В/с
С целью определения реакционной способности образующихся при окислении НР ионов оксоаммония, был смоделирован процесс окисления 4-оксо-2,2,6,6-ТМП (2) ионом оксоаммония (27), путем проведения окисления НР (26) в присутствии амина (2). В результате на ЦВА зафиксировано небольшое снижение пика восстановления иона оксоаммония (27) при 100 кратном избытке амина (2) при снижении скорости развертки потенциала до 0,02 В/с (рисунок 14).

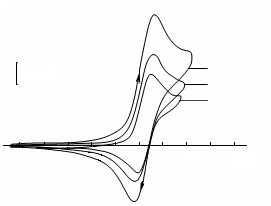
Рисунок 14 – ЦВА 3·10-3 М 4-оксо-2,2,6,6-ТМП-1-оксила (26) на СУ-аноде в среде СН3CN/ 0,1 M Bu4NBF4 и 3·10-1 М 4-оксо-2,2,6,6-ТМП (2) при разных скоростях развертки потенциала: 1 - 0,2 В/с; 2 – 0,1 В/с; 3 – 0,02 В/с
Рисунок 15 – ЦВА 3·10-3 М 4-оксо-2,2,6,6-ТМП-1-оксила (26) на СУ-аноде в среде СН3CN/ 0,1 M Bu4NBF4 и 2,4·10-2 моль 2-пропанола при разных скоростях развертки потенциала: 1 - 0,2 В/с; 2 – 0,1 В/с; 3 – 0,02 В/с
Этот факт подтверждает протекание медленной редокс-реакции между ионом оксоаммония (27) и амином (2). В главе 4 (раздел 4.2, рисунок 7 а, с.15) было показано, что при проведении процесса окисления амина (2) на обратной ветви ЦВА-кривой фиксируется пик ре-восстановления при потенциалах редокс-пары НР (26)/ион оксоаммония (27) (Ep,red = +0,40 В). Интенсивность этого пика возрастает при введении в раствор НР (26). Следовательно, окисление амина (2) непосредственно на электроде приводит к образованию иона оксоаммония (27). Можно полагать, что так же с образованием иона оксоаммония (27) протекает и процесс медиаторного (НР (26)) и непосредственного (на электроде) окисления 4-оксо-2,2,6,6-ТМП (2).
Отдельно было изучено взаимодействие иона оксоаммония (27) с этиловым и изопропиловым спиртами, при контроле методом ЦВА процесса окисления НР (26) в отсутствии и в присутствии этих спиртов. В отличие от пиперидина (2) введение даже 1000 кратного избытка изопропанола слабо отражается на ЦВА-кривых радикала (26) (рисунок 15), что означает малую скорость межмолекулярного электронного переноса между молекулой спирта и ионом оксоаммония (27). Данные ЦВА свидетельствуют о том, что спирты реагируют с ионами оксоаммония, образующимися на аноде, медленно. В то же время известно, что химическое взаимодействие оксоаммониевой соли и спирта протекает относительно быстро, даже при низкой (0 0С) температуре. Этот факт говорит о том, что поведение частиц, находящихся на границе раздела фаз электрод-раствор отличается от их поведения в растворе.
Глава 6. Электрохимическое окисление спиртов с применением каталитической системы нитроксильный радикал (НР) ряда 2,2,6,6-тетраметилпиперидина (2,2,6,6-ТМП) –
йодид калия и 1-галогенпроизводных ряда 2,2,6,6-ТМПСелективное окисление спиртов до альдегидов является актуальной проблемой органического синтеза. Хотя известно множество методов окисления, часто трудно найти метод, который позволяет провести селективное окисление спиртов с применением доступных реагентов и простых способов выделения продуктов реакции. Оксоаммониевые катионы, образующиеся при окислении НР ряда 2,2,6,6-ТМП, окисляют первичные и вторичные спирты до соответствующих карбонильных соединений, а образующийся гидроксиламин окисляется до НР, т.о. каталитический цикл замыкается (схема 8).

Схема 8 – Окисление спиртов с использованием НР в качестве катализаторов
Известно, что при электрохимическом окислении спиртов в присутствии только йодида калия первичные спирты окисляются до эфиров кислот, а вторичные – до соответствующих кетонов. Полагают, что окислителем в этом случае является ион йодония. Однако образование последнего в водных растворах в результате электрохимического окисления йодид-аниона на аноде маловероятно, так как для этого процесса требуется достижение высокого потенциала (+1,8 В). Возможно, что в качестве первичного окислителя выступают и другие продукты электрохимических превращений йодид-аниона (J2, J).
Предложенный в работе новый электрохимический способ окисления первичных и вторичных спиртов до соответствующих карбонильных соединений с применением двухмедиаторной системы йодид калия – НР (33-35) показывает, что присутствие каталитического количества НР ряда 2,2,6,6-ТМП в электролите, содержащем йодид калия и спирт, меняет направление реакции коренным образом. Первичные спирты в этом случае окисляются только до соответствующих альдегидов. Предложен возможный механизм реакции окисления спиртов системой НР – йодид калия (схема 9), где первичным окислителем является электрогенерируемый йод.

Схема 9 – Механизм реакции окисления спиртов двухмедиаторной системой НР – йодид калия
Реакцию окисления проводили в двухфазной системе органический растворитель – водный раствор йодида калия. В качестве органической фазы использовали хлористый метилен, толуол, четыреххлористый углерод. Существенных различий по выходу продуктов реакции в зависимости от органической фазы не наблюдалось. Строение НР (33-35) также практически не влияло на выход карбонильного соединения. Различия в выходе продуктов не превышали 5 %. Установлено, что скорость реакции окисления спиртов существенным образом зависит от рН среды. В слабощелочной среде (0,1 М NaHCO3) реакция проходит быстрее, чем в нейтральной (таблица 3; опыты 4, 7). Это связано с тем, что спирт в щелочной среде вступает в реакцию окисления в виде алкоксида, который легче взаимодействует с катионом оксоаммония. Окисление спиртов в присутствии меньшего количества НР и йодида калия (таблица 3; опыты 3, 6, 8) приводит к снижению выхода соответствующего карбонильного соединения в расчете на исходный спирт.
Таблица 3 – Электрохимическое окисление спиртов с применением каталитической системы йодид калия – 4-ацетиламино-2,2,6,6-ТМП-1-оксил (33) в двухфазной системе CH2Cl2– водный раствор 0,5 М NaHCO3 (рН 8,6); концентрации реагентов: 0,05 М спирт / 0,005 М (10 мол.%) (33) / 0,01 М (20 мол.%) KI
| № опыта | Спирт | Количество пропущенного электричества (Q, F) | Выход по току, % | |
| 2 | 4 | |||
| Выход а альдегида или кетона (на исходный спирт), % | ||||
| 1 | 2 | 3 | 4 | 5 |
| 1 | Бензиловый спирт | 62,5 | 95 (85)г | 47 |
| 2 | Фенилэтиловый спирт | 60,3 | 90 (75)г | 45 |
| 3 б | Фенилэтиловый спирт | 43,6 | 72 | 36 |
| 4 в | Фенилэтиловый спирт | 23 | 52 | 26 |
| 5 | Циклогексанол | 48,7 | 85 (80)г | 42 |
| 1 | 2 | 3 | 4 | 5 |
| 6 б | Циклогексанол | 30 | 65 | 32 |
| 7 в | Циклогексанол | 25 | 67 | 33 |
| 8 д | Циклогексанол | 27 | 57 | 28 |
| 9 | 1-Бутанол | 42 | 85 | 42 |
| 10 | 1-Пентанол | 46,5 | 89 | 44 |
| 11 | 2-Пропанол | 38,7 | 80 | 40 |
а по данным ГЖХ;
б концентрация реагентов 0,05 М спирт/0,005 М (10 мол.%) (33)/0,005 М (10 мол.%) KI;
в рН электролита 6,86 (буферный раствор Na2HPO4 и KH2PO4);
г выход по выделению;
д концентрация реагентов 0,05 М спирт/0,0005 М (1 мол.%) (33)/0,001 М (2 мол.%) KI
Таким образом, селективное электрохимическое окисление первичных и вторичных спиртов с применением каталитической системы НР – йодид калия приводит к образованию альдегидов и кетонов соответственно с хорошим выходом по веществу и току.
Возможности препаративного использования электрохимического метода окисления спиртов с применением 1-хлор-2,2,6,6-ТМП (5) или 2,2,6,6-ТМП (1) показаны на примере окисления циклогексанола и других спиртов. Для проведения реакции в электролит, содержащий спирт, воду, хлористый метилен и хлорид натрия добавляют соединение (5) или (1) и проводят электролиз в бездиафрагменном электролизере на Pt-аноде, при плотности тока 0,15 А/см2. Хлорамин (5) или амин (1) используют в каталитических количествах (5-10 мол. %). При этом окисление спирта до циклогексанона проходит с высоким выходом по веществу и току. После пропускания 3 F электричества электролит по данным ГЖХ содержит 97 % циклогексанона; 1,5 % циклогексанола и 1,5 % НР (13). Если не вводить в электролит хлорамин (5) или амин (1), то окисление циклогексанола практически не происходит. Как оказалось, непрямое электрохимическое окисление первичных и вторичных спиртов с использованием галогенаминов (5), (9) или амина (1) приводит к образованию альдегидов и кетонов с высоким выходом по веществу (65-95 %).
Глава 7. Электрохимическое галогенирование 4-оксо-2,2,6,6-тетраметилпиперидина (ТАА) в кислой среде
7.1 Электрохимическое хлорирование ТАА
Хлорирование ТАА (2) ранее практически не изучалось. Известно только, что монохлорпроизводные ТАА можно получить при действии хлористого сульфурила на раствор гидрохлорида ТАА в хлористом метилене. В процессе изучения реакции электрохимического хлорирования ТАА (2) установлено, что состав продуктов электролиза можно регулировать, и в зависимости от условий электролиза могут быть получены 3-хлор-4-оксо-2,2,6,6-ТМП (36), 3,5-дихлор-4-оксо-2,2,6,6-ТМП (37), 3,3,5,5-тетрахлор-4-оксо-2,2,6,6-ТМП гидрохлорид (38) и 1-хлор-4-оксо-2,2,6,6-ТМП (6) (схема 10). Попытка получить в аналитически чистом виде (36) и (37) не удалась, так как в результате синтеза образовывались смеси, содержащие моно-, ди- и тетрахлорзамещенные производные ТАА (2). Хотя содержание примесей было незначительным, они кристаллизовались совместно с основным веществом. Анализ смеси соединений (36) и (37) был проведен путем их превращения в амиды (39) и (40) при обработке аммиаком. Состав смеси амидов определяли спектрофотометрическим методом. При восстановлении смеси амальгамой натрия образуется амид (39), который используется для синтеза спиновых меток. 4-Оксо-3,3,5,5-тетрахлор-2,2,6,6-ТМП обладает очень слабыми основными свойствами, и его хлороводородная соль (38) при действии воды полностью гидролизуется с образованием амина (41). Строение 4-оксо-3,3,5,5-тетрахлор-2,2,6,6-тетраметилпиперидина (41) подтверждено данными элементного анализа, ИК- и ПМР-спектроскопии.

Схема 10 - Электрохимическое хлорирование ТАА (2) в кислой среде
В таблице 4 приведены данные о влиянии условий электролиза на состав и выход продуктов реакции. Как видно из данных таблицы выход продуктов электролиза и состав смеси зависит от концентрации соляной кислоты, от количества пропущенного электричества и материала анода.
Таблица 4 - Влияние условий электролиза на выход продуктов реакции электрохимического хлорирования ТАА (2 ) в кислой среде; плотность тока 0,1 А/см2
| Материал электрода | Диаф-рагма | Темпера-тура, оС | Концен-трация HCl, М | Q, F | Продукты, выход по веществу, % * | |||
| 38 | 36 | 37 | 6 | |||||
| Платина ** | + | 20 | 3 | 4 | – | – | – | 57 |
| Платина | + | 20 | 6 | 4 | – | 42 | 18 | – |
| Платина | + | 20 | 6 | 8 | 55 | 20 | 10 | – |
| Платина | + | 20 | 12 | 4 | 34 | 24 | 6 | – |
| Платина | + | 20 | 12 | 8 | 70 | – | – | – |
| Платина | + | 50 | 6 | 4 | – | 51 | 18 | – |
| Платина | – | 50 | 6 | 4 | – | 39 | 27 | – |
| Платина | + | 50 | 6 | 2 | – | 6 | 49 | – |
| Платина | + | 50 | 12 | 4 | – | 65 | 14 | – |
| Платина | + | 50 | 12 | 8 | 68 | – | – | – |
| Платина | – | 50 | 12 | 8 | 67 | – | – | – |
| Платина | + | 70 | 12 | 8 | 23 | – | – | – |
| Графит | + | 20 | 12 | 8 | 19 | 40 | 10 | – |
| Графит | + | 50 | 12 | 8 | 60 | - | - | - |
| СУ | + | 50 | 12 | 8 | - | 18 | 7 | |
* состав смеси соединений (36) и (37) был определен путем их превращения в соответствующие 3-карбоксамидо-2,2,5,5-тетраметилпирролидин (39) и 3-карбоксамидо-2,2,5,5-тетраметилпирролин (40). Соединение (6) было выделено и охарактеризовано;
** синтез проведен в двухфазной системе в присутствии хлористого метилена
Значительное влияние на процесс электрохимического галогенирования ТАА (2) оказывает концентрация соляной кислоты. При проведении электролиза в 3 М соляной кислоте в диафрагменном или бездиафрагменном электролизере, при температуре 20 0С и плотности тока 0,1 А/см2 основным продуктом реакции является соединение (6). При увеличении концентрации кислоты до 6 М образуется смесь (36) и (37). Электрохимическое хлорирование ТАА (2) в концентрированной соляной кислоте (12 М) приводит к образованию продукта полного хлорирования (38). Селективность процесса хлорирования ТАА (2), возможно, связана с понижением реакционной способности 3-хлор-4-оксо-2,2,6,6-ТМП (36) по сравнению с ТАА (2). Вероятно, это связано с тем, что в концентрированной кислоте изменяется стадия, определяющая скорость реакции (енолизация – в разбавленной соляной кислоте, присоединение хлора – в концентрированной кислоте).
Изучение механизма процесса хлорирования ТАА (2) методом ЦВА показало, что соединение (2) не является деполяризатором по отношению к Pt или графитовому анодам, так как введение его в раствор (5·10-3 М) затрудняет электрохимический процесс и сдвигает потенциал окисления хлорид-иона от 0,9 до 1,0 В. Вероятно, это связано с адсорбцией вещества (2) на аноде. Таким образом, на аноде происходит только окисление хлорид-аниона, что наблюдается в области разряда фона, с дальнейшим образованием молекулярного хлора, а процесс хлорирования идет в растворе. Сам ТАА (2) окисляется при достаточно высоких потенциалах, значительно превышающих потенциал разряда фона и окисления хлорид-анионов. Это подтверждается данными ЦВА раствора ТАА (2) (5·10-3 М) в хлорной кислоте на Pt-аноде. В этом случае наблюдается волна окисления ТАА (2) при потенциале 2,1 В, что значительно превышает потенциал выделения хлора из 10 %-ной соляной кислоты в присутствии (2) (1,1 В). Окисление ТАА (2) является, по-видимому, причиной низкого выхода продуктов хлорирования при плохом перемешивании электролита.
7.2 Электрохимическое бромирование 4-оксо-2,2,6,6-тетраметилпипе-ридина (ТАА)
7.2.1 Электрохимическое бромирование ТАА в бромистоводородной кислоте
Наибольшее значение среди других галогенпроизводных ТАА (2) имеет 3,5-дибром-4-оксо-2,2,6,6-тетраметилпиперидин (42). Его используют для получения НР ряда 2,2,5,5-тетраметилпирролина и 2,2,5,5-тетраметилпирролидина. Соединение (42) может быть получено бромированием ТАА (2) бромом в растворе уксусной кислоты при 20 0С или в растворе бромистоводородной кислоты при 80-90 0С.
В работе показано, что удобным методом получения гидробромида 3,5-дибром-4-оксо-2,2,6,6-ТМП (43) является электрохимическое бромирование ТАА (2) в бромистоводородной кислоте на Pt-аноде. Метод прост в исполнении и позволяет получать (43) с выходом более 90 % в широком интервале условий. Электрохимическое бромирование ТАА (2) проходит избирательно. Реакция останавливается на стадии образования соли (43), которая практически не растворима в бромистоводородной кислоте. Выбор условий электролиза основан на данных препаративного электролиза и ЦВА. Решающее влияние на процесс бромирования оказывает температура. При 20 0С реакция бромирования ТАА (2) идет медленно. Образующийся на аноде бром не успевает полностью прореагировать с ТАА (2). Для препаративного электролиза необходимо поддерживать температуру в интервале 60-70 оС, так как в этом случае процесс можно провести достаточно быстро с приемлемой объемной плотностью тока (0,01 А/см3). Силу тока необходимо регулировать таким образом, чтобы в реакционной смеси не накапливался бром. Изменение плотности тока в пределах 0,02-0,10 А/см2 не оказывает существенного влияния на выход 3,5-дибром-4-оксо-2,2,6,6-ТМП гидробромида (43). Реакцию можно провести в бездиафрагменном электролизере, но в этом случае выход по току снижается на 15–20 %. При использовании графитового анода выход соединения (43) снижается на 5-10 %. Графит в процессе синтеза постепенно разрушается и загрязняет выпадающий в осадок гидробромид (43). Для электролиза используют раствор ТАА (2) в бромистоводородной кислоте при соотношении мольных концентраций 1:4 соответственно. В конце электролиза бромистоводородная кислота должна иметь концентрацию не ниже, чем 1,0-1,5 М, так как гидробромид (43) в водном растворе при нагревании подвергается гидролизу с образованием 3,4-диоксо-2,2,6,6-ТМП. Кроме этого при снижении концентрации бромистоводородной кислоты возможно окисление брома до бромноватой кислоты.
С целью установить механизм электрохимического бромирования ТАА (2), процесс был изучен методом ЦВА. Согласно анализу данных ЦВА добавка ТАА (2) к раствору бромистоводородной кислоты не влияет на потенциал Pt-анода, при котором происходит разряд бромид-анионов. На основании этого можно считать процесс бромирования ТАА (2) непрямым. Кроме этого процесс окисления ТАА (2) на Pt происходит при более положительном потенциале, чем окисление бромид-анионов. Анализ зависимости величины Ipa и Ipk от скорости развертки потенциала показывает, что бромирование ТАА (2) проходит по механизму ЕС. При низких скоростях развертки потенциала Ipa > Ipk, а при увеличении скорости развертки Ipk Ipa. Скорость электрохимического процесса окисления бромид-аниона возрастает с увеличением температуры, растет величина пика анодного тока, то есть повышение температуры способствует увеличению скорости как химической реакции бромирования ТАА (2), так и электрохимической реакции разряда бромид-аниона.
При электрохимическом бромировании 2,2,6,6-ТМП (1) и 4-гидрокси-2,2,6,6-ТМП (3) (соединений, не содержащих карбонильной группы) получены пербромиды соответствующих гидробромидов. Сходство потенциодинамических кривых разряда бромид-аниона в присутствии ТАА (2), 2,2,6,6-ТМП (1) или 4-гидрокси-2,2,6,6-ТМП (3) свидетельствует о том, что процесс бромирования этих соединений проходит по одному и тому же механизму, то есть первоначальным продуктом реакций бромирования указанных соединений является образование соответствующих пербромидов. Таким образом, реакция ТАА (2) с электрохимически генерируемым бромом ведет к получению соединения (43) через образование пербромида (44), что представлено на схеме 11:

Схема 11 - Электрохимическое бромирование ТАА (2)
Попытка провести электрохимическое бромирование 4-оксо-2,2,6,6-ТМП-1-оксила (26) не удалась, так как бром мгновенно реагирует с нитроксильной группой с образованием соответствующей высокореакционноспособной оксоаммониевой соли (ОС).
7.2.2 Электрохимическое бромирование 4-оксо-2,2,6,6-тетраметилпиперидина (ТАА) в электролите бромид калия – серная кислота – вода
С целью использовать в препаративном синтезе вместо бромистоводородной кислоты более доступный и удобный реагент – бромид калия, изучен процесс электрохимического бромирования ТАА (2) в электролите бромид калия – серная кислота – вода (схема 12).

Схема 12 - Электрохимическое бромирование ТАА (2) в электролите бромид калия – серная кислота – вода
Синтез проводили в условиях, аналогичных электрохимическому бромированию ТАА (2) в бромистоводородной кислоте. Оказалось, что в этом случае выход гидробромида (43) не превышает 25-30 %. В связи с этим было изучено электрохимическое поведение этой системы. Синтез, проведенный в гальваностатическом режиме, показал, что процесс бромирования ТАА (2) проходит при постоянном потенциале (0,78-0,80 В), который не изменяется в процессе всего синтеза (количество пропущенного электричества 4 F). При этом потенциале происходит только окисление бромид-ионов, а ТАА (2) не окисляется. Это подтверждается также поляризационными измерениями. Введение в электролит ТАА (2) практически не влияет на вид поляризационной кривой (рисунок 16). Таким образом, снижение выхода гидробромида (43) связано либо с его разложением, либо с восстановительными процессами, происходящими на катоде.
Рисунок 16 – Поляризационные кривые: 1 – фон: 0,67 М КВr + 0,4 М (60 мл) H2SO4 + 40 мл Н2О; 2 - фон + 10-4 М ТАА (2); 3 – фон + 10-3 М ТАА (2); анод - Pt
Препаративный синтез в диафрагменном электролизере показал правильность последнего предположения, выход гидробромида (43) в этом случае составил 80 %. Кроме этого, при восстановлении соединения (43) на Pt-катоде в растворе серной кислоты основным продуктом реакции является ТАА (2), а на свинцовом катоде образуется сложная смесь продуктов восстановления (по данным ГЖХ), включающая ТАА (2), 4-гидрокси-2,2,6,6-ТМП (3), 2,2,6,6-ТМП (1). Восстановление соединения (43) в растворе бромистоводородной кислоты не происходит, вероятно, из-за низкой растворимости.
Зависимость выхода гидробромида (43) от условий проведения электролиза представлена в таблице 5.
Таблица 5 - Зависимость выхода гидробромида 3,5-дибром-4-оксо-2,2,6,6-ТМП (43) от условий электролиза (анод и катод – Pt-пластинки площадью 10 см2, t = 60 оС, количество пропущенного электричества - 4 F)
| Номер опыта | Соотношение исходных реагентов, ТАА(2) : KBr : H2SO4,М | j, А/см2 | Наличие диафрагмы | Выход по веществу *,% |
| 1 2 3 4 5** 6*** 7 | 0,1 : 0,25 : 0,2 0,1 : 0,40 : 0,2 0,1 : 0,40 : 0,2 0,1 : 0,40 : 0,2 0,1 : 0,40 : 0,2 0,1 : 0,40 : 0,2 0,1 : 0,50 : 0,2 | 0,1 0,1 0,1 0,05 0,2 0,1 0,1 | + + - + + + + | 55 78 21 74 39 75 80 |
* Полученный в результате электролиза продукт содержал кроме соединения (43) 0,73 % гидросульфата калия;
** Cинтез проведен при температуре 30 оС. При его проведении образуется пербромид ТАА (2), который постепенно переходит в гидробромид 3,5-дибром-4-оксо-2,2,6,6- ТМП (43);
***Температура электролита 80 оС
Как видно из данных таблицы, процесс может быть реализован только в диафрагменном электролизере. Снижение выхода соединения (43) при проведении электролиза в бездиафрагменном электролизере связано с восстановлением продукта на катоде (данные препаративного электролиза и ГЖХ).
Глава 8 Электрохимический синтез соединений ряда 2,2,5,5-тетраметилпирролидина
8.1 Электрохимическое иодирование 4-оксо-2,2.6.6-тетраметилпи
перидина (ТАА) в электролите йодид калия -гидроксид калия -водный раствор аммиака. Синтез 3-карбоксамидо-2,2,5,5-тетраметилпирролидинаЭлектрохимическое иодирование ТАА (2) представляет особый интерес, так как открывает путь к одностадийному синтезу производных 2,2,5,5-тетраметилпирролидина – важнейших исходных веществ для получения НР этого ряда и спиновых меток в частности. Каталог фирмы “Aldrich” предлагает более 50 наименований НР, половина из которых представляют собой соединения ряда 2,2,5,5-тетраметилпирролидина.
В работе показано, что синтез 3-карбоксамидо-2,2,5,5-тетраметилпирролидина (39) может быть осуществлен электрохимически в бездиафрагменном электролизере на Pt-аноде. Общая схема синтеза представлена ниже (схема 13). Электрохимический вариант синтеза 3-карбоксамидо-2,2,5,5-тетраметилпирролидина (39) позволяет провести перегруппировку Фаворского без расходования йода и в практически неизменяющихся условиях. Образующийся побочно в процессе электрохимического йодирования в щелочной среде KIO3 восстанавливается на катоде до KI. Таким образом, в процессе иодирования ТАА (2) в бездиафрагменном электролизере концентрация KI существенно не изменяется.

Схема 13 - Электрохимическое иодирование ТАА (2). Электрохимический синтез 3-карбоксамидо-2,2,5,5-тетраметилпирролидина (39) и 3-карбоксамидо-2,2,5,5-тетраметилпирролина (40)
Электролиз проводили в электролите ТАА (2) – KI – KOH - NH3 на Pt-аноде при температуре 20 - 40 оС. Выход продуктов реакции cоставил 70-75 % по веществу и 15-30 % по току после их выделения из реакционной смеси. Иодпроизводные (45) и (46) в условиях реакции не выделяются и в момент образования в результате перегруппировки Фаворского превращаются в соответствующие амиды (39) и (40).
Механизм процесса электрохимического иодирования ТАА (2) изучен методом ЦВА. В системе КI - KOH - NH3 процесс электрохимического образования йода происходит при потенциале 0,2 В и является необратимым. При увеличении скорости развертки потенциала катодный пик увеличивается. При введении в электролит ТАА (2) процесс становится полностью необратимым, катодный пик исчезает. Эти данные подтверждают механизм ЕС (A - e B C), то есть быструю химическую реакцию, следующую за переносом электрона.
Реакция электрохимического иодирования ТАА (2) исследовалась также препаративными методами. Выход продуктов реакции определяли после их выделения из реакционной смеси. Полученные данные о влиянии условий электролиза на выход продуктов реакции и их состав представлены в таблице 6. Данные таблицы показывают, что хороший выход по веществу может быть достигнут в широком диапазоне условий. Выход по току значительно увеличивается при проведении электролиза в диафрагменном электролизере. Однако в этом случае анолит надо регенерировать, так как в процессе электролиза в анолите накапливается иодат калия.
В электрохимическом синтезе образуется смесь соединений (39) и (40), которые разделить кристаллизацией или возгонкой не удается. Для того, чтобы можно было использовать этот метод получения 3-карбоксамидо-2,2,5,5-тетраметилпирролидина (39) в препаративных целях была исследована реакция электрохимического восстановления 3-карбоксамидо-2,2,5,5-тетраметилпирролина (40). Эта реакция приводит к восстановлению двойной связи в пирролине (40) и образованию с количественным выходом соответствующего пирролидина (39). Реакция электрохимического восстановления пирролина (40) детально рассматривается в разделе 8.3.
Таблица 6 - Влияние условий электролиза на выход смеси соединений (39) и (40) в реакции электрохимического иодирования ТАА (2)
| № п/п | Состав электролита ТАА(2):КОН:KI, моль/л | j, A/ см2 | Q,F | Диа-фраг-ма | Кон-вер-сия | Выход по ве-ществу, %** | Содержание в продукте реакции *, % | |
| соединения (39) | соединения (40) | |||||||
| 1 2 3 4*** 5*** 6 7 8 | 0,5:1:0,5 0,5:2:0,5 0,5:2:0,5 0,5:2:0,5 0,5:2:0,5 0,5:2:0,5 0,5:2:0,5 0,5:2:0,5 | 0,1 0,1 0,1 0,1 0,1 0,1 0,1 0,5 | 4 2 4 4 8 2 4 4 | - - - - - + + - | 28 20 50 60 64 42 89 48 | 64 76 70 55 65 78 70 75 | 95 93 92 87 85 93,5 93,5 97 | 5 7 8 13 15 6,5 6,5 3 |
* по данным ГЖХ;
**выход рассчитан, исходя из того, что основным продуктом реакции является соединение (39);
***синтез проведен в двухфазной системе в присутствии бензола
8.2 Электрохимическое галогенирование 4-оксо-2,2,6,6-тетраметилпи
перидина (ТАА) в растворе метилата (или этилата) натрияВ работе предложен метод синтеза производных 3-карбметокси-2,2,5,5-тетраметилпирролидина (47). Для этого была изучена реакция электрохимичекого галогенирования ТАА (2) в электролите КВг (КI, КС1) - СН3ОNа. Показано, что галогенпроизводные ТАА (2) в присутствии метилата натрия в момент образования подвергаются перегруппировке Фаворского с образованием соответствующих эфиров кислот (47) и (48). Вероятно, промежуточными продуктами в этом синтезе являются моно- и дигалогенпроизводные ТАА (2) (схема 14).

Схема 14 - Электрохимическое галогенирование ТАА (2) в растворе метилата натрия
Синтез эфиров проводят в бездиафрагменном электролизере, снабженном обратным холодильником и электродным пакетом: анод –Pt-пластинка, катод – две пластины из коррозионностойкой стали, расстояние между электродами 3 мм, при плотности тока 0,1 А/см2, температуре 30-40 0С, перемешивании и заканчивают после пропускания 8 F электричества. Состав смеси эфиров (47) и (48) определяют с помощью ГЖХ и спектрофотометрического метода (таблица 7).
Таблица 7 - Влияние условий электролиза на выход 3-карбметокси-2,2,5,5-тетраметилпирролидина (47) и 3-карбметокси-2,2,5,5-тетраметилпирролина (48) в реакции электрохимического галогенирования ТАА (2) в растворе метилата натрия
| № опыта | Состав электролита, (М) | Q, F | Выход смеси соединений (47) и (48), %.* | Состав смеси по данным ГЖХ, % | Состав смеси по данным УФ-спектров, % ** | ||||
| (2) | (47) | (48) | (2) | (47) | (48) | ||||
| 1 | ТАА(2)-КCl-CH3ONa 1 : 1,7 : 2,6 | 2 | 86 | 79 | 7 | 14 | - | - | - |
| 2 | - // - | 4 | 84 | 38 | 12 | 50 | - | - | - |
| 3 | - // - | 8 | 80 | 0 | 16 | 84 | 0 | 15 | 85 |
| 4 | ТАА(2)-КBr-CH3ONa 1 : 1,7 : 2,6 | 2 | 87 | 76 | 6 | 18 | - | - | - |
| 5 | - // - | 4 | 85 | 28 | 20 | 52 | - | - | - |
| 6 | - // - | 8 | 85 | 0 | 38 | 62 | 0 | 40 | 60 |
| 7 | ТАА(2)-КI-CH3ONa 1 : 1,7 : 2,6 | 2 | 84 | 82 | 8 | 10 | - | - | - |
| 8 | - // - | 4 | 81 | 31 | 30 | 39 | - | - | - |
| 9 | - // - | 8 | 82 | 0 | 46,8 | 53,2 | 0 | 48 | 52 |
* выход смеси соединений на вступивший в реакцию ТАА (2);
** для опытов 1, 2, 4, 5, 7, 8 этот метод анализа не использовался, т.к. в смеси оставался ТАА (2)
Согласно данным, приведенным в таблице, независимо от природы галогенид-иона выход смеси продуктов реакции составил 80-87 % и зависел от количества пропущенного электричества. При пропускании 8 F электричества происходило полное превращение исходного вещества ТАА (2), что значительно упрощало выделение продуктов реакции. Количественный анализ смеси 3-карбметокси-2,2,5,5-тетраметилпирролина (48) и 3-карбметокси-2,2,5,5-тетраметилпирролидина (47) проведен спектрофотометрическим методом. Спектры поглощения чистых веществ и их смеси представлены на рисунке 17.
Рисунок 17 - УФ-спектры поглощения соединений (47) и (48) (С = 5·10-5 М) в метаноле: 1 – 3-карбметокси-2,2,5,5-тетраметилпирролин (48)*; 2 – 3-карбметокси-2,2,5,5-тетраметилпирроли-дин (47)*; 3 – смесь продуктов (47) и (48)**;
* получен по известной методике; ** cмесь получена в результате электрохимического синтеза
Расчет проводили, исходя из допущения, что насыщенный эфир (47) прозрачен – молярный коэффициент поглощения () для 3-карбметокси-2,2,5,5-тетраметилпирролидина (47) равен при 204 нм 980, а для 3-карбметокси-2,2,5,5-тетраметилпирроллина (48) при этой же длине волны – 12800.
Процесс электрохимического превращения ТАА (2) в растворе метилата натрия и йодида калия изучен методом ЦВА. Показано, что окисление I– происходит при потенциале 0,20 В и является необратимым. Добавка ТАА (2) к электролиту, содержащему CH3ONa – KI, приводит к исчезновению катодного пика и смещению потенциала окисления I– от 0,2 В до 0,6 В, таким образом, реакция окисления I– становится полностью необратимой, а процесс иодирования ТАА (2) является непрямым. При увеличении скорости развертки потенциала значение потенциала анодного пика смещается в положительную область, увеличивается величина тока этого пика. При низких скоростях развертки потенциала катодный пик практически исчезает, а при увеличении скорости развертки потенциала катодный пик растет, что подтверждает механизм ЕС для процесса иодирования ТАА (2), т.е. быструю химическую реакцию, следующую за переносом электрона (А - е В С).
Аналогично электрохимическому галогенированию ТАА (2) в растворе метилата натрия проведены реакции в растворе этилата натрия. В результате была также получена смесь соединений 3-карбэтокси-2,2,5,5-тетраметилпирролидина (49) и 3-карбэтокси-2,2,5,5-тетраметилпирролина (50).
8.3 Электрохимическое восстановление соединений ряда 2,2,5,5-тетраметилпирролина до соединений ряда 2,2,5,5-тетраметилпир
ролидина на ртутном катодеВ электрохимическом синтезе 3-карбоксамидо-2,2,5,5-тетраметилпирролидина (39) образуется смесь соединений, содержащая главным образом, пирролидин (39), (выход по веществу 70 – 75 %, по току 25 – 30 %) с примесью соединения (40). Хотя примесь этого соединения незначительна (от 3 до 15 %) отделить ее от основного продукта кристаллизацией или возгонкой не удается. Для того, чтобы можно было использовать этот метод получения 3-карбоксамидо-2,2,5,5-тетраметилпирролидина (39) в препаративных целях изучена реакция электрохимиического восстановления на амальгаме натрия 3-карбоксамидо-2,2,5,5-тетраметилпирролина (40). Эта реакция приводит к восстановлению двойной связи и образованию с количественным выходом соответствующего пирролидина (39).
Аналогичным образом из 3-карбоксамидо-2,2,5,5-тетраметилпирролин-1-оксила (51) получен восстановлением на ртутном катоде 3-карбоксамидо-2,2,5,5-тетраметилпирролидин-1-оксил (52). Предложенный электрохимический метод восстановления двойной связи в радикале (51) является, по-видимому, наиболее простым и удобным (схема 15), так как применение других методов ограничено из-за восстановления нитроксильной группы до амина. Образующийся в результате электрохимического процесса 3-карбоксамидо-1-гидрокси-2,2,5,5-тетраметилпирролидин (53) легко с количественным выходом окисляется в соответствующий НР (52).

Схема 15 - Восстановление двойной связи в 3-карбоксамидо-2,2,5,5-тетраметилпирролин-1-оксиле (51) на ртутном катоде
*- окисление можно провести кислородом воздуха
Как видно на рисунках 18 и 19 восстановление нитроксильной группы является прямым процессом, а восстановление двойной связи происходит амальгамой, образующейся при электролизе раствора NaОН на ртутном катоде. При проведении процесса восстановления при потенциале 0,80 В, когда образование амальгамы исключено, единственным продуктом реакции является соединение (53).
Рисунок 18 - Поляризационные кривые восстановления 3-карбоксамидо-2,2,5,5-тетраметилпирролин-1-оксила (51). Катод - ртуть, анод – платина; 1- фон: 7 %-ый NaOH; 2- фон/10-4 М соединения (51); 3 - фон/210-4 М соединения (51); = 0,002 В/с; t = 20 оС
Рисунок 19 - Поляризационные кривые восстановления 3-карбоксамидо-2,2,5,5-тетраметилпирролина (40). Катод - ртуть, анод – платина; 1- фон: 7 %-ый NaOH; 2- фон/10-4 М соединения (40); 3 – фон/210-4 М соединения (40); = 0,002 В/с; t= 20 оС
Разработанный метод восстановления двойной углерод-углерод связи в смеси соединений ряда пирролина и пирролидина на амальгаме натрия был применен также для получения эфиров (47, 49) ряда 2,2,5,5-тетраметилпирролидина. Использовать прямое электрохимическое восстановление двойной связи в соединениях (48) и (50) провести не удалось, так как смесь эфиров не растворима в воде. Поэтому восстановление проводили в этаноле на предварительно полученной электролизом раствора NaOH амальгаме натрия. Выход пирролидинов (47) и (49) при восстановлении пирролинов (48) и (50) на амальгаме натрия количественный. Происходящие превращения представлены на схеме 16.

Схема 16 - Восстановление двойной связи в 3-карбметокси-2,2,5,5-тетраметилпирролидине (48) и 3-карбэтокси-2,2,5,5-тетраметилпирролидине (50)
Предложенный метод восстановления пирролинов до пирролидинов существенно повышает перспективы использования одностадийного метода получения производных ряда 2,2,5,5-тетраметилпирролидина.
Глава 9 Использование галогенпроизводных ряда 2,2,6,6-тетраметилпиперидина (2,2,6,6-ТМП) в органическом синтезе
9.1 Применение 1-галогенпроизводных ряда 2,2,6,6-ТМП в качестве дегидрирующих и галогенирующих агентов ароматических соединений
Изучение новых реагентов для окисления фенолов является важной задачей вследствие разнообразия окислительных реакций фенолов и зависимости результатов от строения субстрата и окислителя. Исследовалась возможность использования 1-галогенпроизводных ряда 2,2,6,6-ТМП в качестве окислителей и галогенирующих агентов. Полученные результаты показали, что галогенамины ряда 2,2,6,6-ТМП имеют хорошую перспективу использования в реакциях окисления фенолов.
Направление реакции окисления фенолов 1-галогенпроизводными ряда 2,2,6,6-ТМП в значительной мере определяется строением фенола. 2,6-Ди-трет-бутил-4-метилфенол (54) окисляется 1-хлор- (7) или 1-бром-4-гидрокси-2,2,2,6-ТМП (11) в нейтральной среде (рН 6-7) с образованием 3,3’,5,5’-тетра-трет-бутилстильбенхинона (55):

При окислении в нейтральной среде (рН 6-7) 2,6-ди-трет-бутилфенола (56) 1-хлор- (5) или 1-бром-2,2,6,6-ТМП (9) также происходит димеризация и образование 3,3',5,5'-тетра-трет-бутилдифенохинона (57). В органическом растворителе, по-видимому, реакция димеризации проходит по радикальному механизму. Скорость реакции зависит от скорости протонирования N-галогенпроизводного. Стабильность образующегося феноксильного радикала определяется структурными особенностями молекулы исходного фенола.

При окислении 2,4,6-три-трет-бутилфенола (58) из реакционной среды (рН 6-7) были выделены 2,6-ди-трет-бутилбензохинон-1,4 (59) и исходный фенол (58). 2,4,6-Три-трет-бутилфенол (58) практически не подвергается димеризации, вследствие высокой стабильности образующегося из него радикала - 2,4,6-три-трет-бутилфеноксила. 1-Хлор-2,2,6,6-ТМП (5) в органическом растворителе окисляет гидрохинон (60) до п-бензохинона (61):

Известно, что феноксильные радикалы с объемными заместителями в 2,4,6-положениях не склонны к С-С димеризации, но достаточно легко взаимодействуют с кислородом с образованием органических пероксидов. Последние легко расщепляются по О-О связи, образуя новые радикалы, которые стабилизируются путем превращения в орто- или пара- бензохиноны. При этом происходит отщепление заместителя. Именно этим, вероятно, объясняется образование хинона (59). -Нафтол (62), взаимодействуя с 1-хлорпроизводными ряда 2,2,6,6-ТМП в гомогенной среде, превращается в 1-хлор-2-нафтол (63) и продукты окисления, среди которых хроматографически обнаружен 1,2-нафтохинон. Фенол (64) при взаимодействии с 1-хлорпроизводными ряда 2,2,6,6-ТМП образует 2,4,6-трихлорфенол (65 а), а c 1-бромпроизводными - 2,4,6-трибромфенол (65 б):

В кислой среде (рН 1-3), по-видимому, реакции протекают по электрофильному механизму, происходит галогенирование субстрата. Такой механизм подтверждается взаимодействием ацетанилида (66) с 1-хлор- (5) или 1-бром-2,2,6,6-ТМП (9). В данном случае образуется единственный продукт - п-хлор- (67 а) или п-бромацетанилид (67 б):

N-Галогенпроизводные ряда 2,2,6,6-ТМП также реагируют с диметилфениламином (68), диэтилфениламином (69) с образованием п-галогенпроизводных этих ароматических аминов. В кислой среде (рН 1-3) 2,4,6-три-трет-бутилфенол (58) взаимодействует с 1-галогенпроизводными 2,2,6,6-ТМП с образованием продукта присоединения в пара-положение кольца - 2,4,6-три-трет-бутил-4-галогенгексадиенона (70 а, б).

Подобные структуры являются результатом диспропорционирования феноксильного радикала под действием хлористо- и бромистоводородных кислот. Строение всех полученных соединений подтверждено данными элементного анализа, ПМР-спектроскопии и сравнением с заведомыми образцами. Все синтезированные структуры образуются с выходом 80-90 %. Соединения (59) и (63) получаются с выходом 50-55 %.
9.2 Синтезы на основе галогенпроизводных 4-оксо-2,2,6,6-тетраметилпиперидина (ТАА)
В работе на основе 3,3,5,5-тетрахлор-4-оксо-2,2,6,6-ТМП (41) (глава 7) были проведены превращения, показанные на схеме 17.

Схема 17 – Химические превращения 3,3,5,5-тетрахлор-4-оксо-2,2,6,6-ТМП (41)
При восстановлении соединения (41) получен соответствующий спирт (71), а при действии аммиака на (41) вследствие перегруппировки Фаворского образуется 3-гидрокси-3-карбоксамидо-4-оксо-2,2,5,5-тетраметилпирролидин (72).
При электрохимическом синтезе гидробромида 3,5-дибром-4-оксо-2,2,6,6-ТМП (43) наблюдалось образование побочных продуктов, связанное с его разложением, поэтому были изучены некоторые свойства этого соединения. Так при кипячении гидробромида (43) с водой с выходом 50 % образуется дикетон (73). Дикетон (73) был восстановлен боргидридом натрия в соответствующий диол (74) (схема 18). Дикетон (73) и диол (74) системой перекись водорода-вольфрамат натрия окислить в соответствующие НР не удалось.

Схема 18 - Химические превращения 3,5-дибром-4-оксо-2,2,6,6-ТМП гидробромида (43)
Для получения НР ряда 2,2,5,5-тетраметилпирролина изучено электрохимическое бромирование 4-оксо-2,2,6,6-ТМП-1-оксила (26). Так как НР неустойчивы в кислой среде, то радикал (26) был превращен в соль соответствующего гидроксиламина (75). Оказалось, что при электрохимическом бромировании (75) в растворе бромистоводородной кислоты происходят глубокие деструктивные изменения, и образуется смесь соединений, состав которой не изучался. Вследствие того, что 3,5-дибром-4-оксо-2,2,6,6-ТМП-1-оксил (76) является важной спиновой меткой, был разработан метод его получения бромированием гидроксиламина (75) в уксусной кислоте, и изучены некоторые его превращения (схема 19).

Схема 19 - Синтез З,5-дибром-4-оксо-2,2,6,6-ТМП-1-оксила (76)
Следует отметить, что соединение (77), также как некоторые другие пиперидиновые галогензамещенные гидроксиламины не окисляются системой H2O2- Na2WO4.
Возможность осуществления реакций НР без затрагивания парамагнитного центра распространили на получение бирадикала 4,4-бис-4-гидрокси-2,2,6,6-ТМП-1-оксила (79) (схема 20). Это соединение синтезировали с перспективой изучения реакционной способности и условий его полимеризации.

Схема 20 – Синтез бирадикала (79)
При взаимодействии радикала (76) с аминами происходит перегруппировка Фаворского, и образуются производные 3-карбоксамидо-2,2,5,5-тетраметилпирролин-1-оксила. На основе этой реакции при взаимодействии радикала (76) с 4-амино-2,2,6,6-тетраметилпиперидин-1-оксилом (81) был получен бирадикал 4-(3-карбоксамидо-2,5-дигидро-2,2,5,5-тетраметил-1Н-пиррол)-2,2,6,6-тетраметилпиперидин-1-оксил (82). Это соединение было получено также встречным синтезом из 3,5-дибром-4-оксо-2,2,6,6-ТМП (42) (схема 21).
Соединение (82) было использовано в качестве добавки к активной массе в источниках тока. Строение всех полученных соединений подтверждено данными ИК-, ПМР-спектроскопии и элементного анализа.

Схема 21 - Синтез бирадикала (82)
Разработанные методы применяются для получения соединений (71-74, 75, 79, 82) в качестве реактивов в органическом синтезе и биотехнологии.
Заключение
Проведенные комплексные исследования, накопленные новые экспериментальные данные позволили осуществить теоретические обобщения и разработать технологические решения по созданию эффективных электрохимических методов галогенирования и окисления ПЗА, НР ряда 2,2,6,6-ТМП и на их основе новых путей синтеза практически важных соединений ряда 2,2,6,6-ТМП и 2,2,5,5-тетраметилпирролидина. Изучение свойств полученных 1-галогенаминов ряда 2,2,6,6-ТМП показало, что эти соединения можно использовать в качестве галогенирующих и дегидрирующих реагентов, позволяющих проводить реакции органического синтеза в мягких условиях. Кроме этого, впервые на примере окисления спиртов показано, что 1-галогенамины и их предшественники - ПЗА ряда 2,2,6,6-ТМП могут эффективно использоваться в каталитических процессах непрямого электрохимического окисления. Значение технических приложений данной работы подтверждается патентами, актами внедрения результатов работы и утвержденными лабораторными методиками и ТУ на способы получения реактивов 7 наименований.
Выводы
1. Разработаны эффективные электрохимические методы синтеза 1-хлор- и 1-бромпроизводных ряда 2,2,6,6-тетраметилпиперидина, которые образуются с высоким выходом по веществу (80-95 %) и току (75-90 %). Изучена стабильность 1-галогенпроизводных ряда 2,2,6,6-тетраметилпиперидина на примере 1-хлор-2,2,6,6-тетраметилпиперидина и 1-хлор-4-гидрокси-2,2,6,6-тетраметил-пиперидина. Показано, что стабильность этих соединений зависит от их строения и уменьшается в растворах с понижением рН, при облучении УФ-светом и продолжительности хранения.
2. Найдены основные закономерности электрохимических превращений пространственно-затрудненных аминов и галогенаминов ряда 2,2,6,6- тетраметилпиперидина в условиях реакции окисления, что позволило впервые разработать и предложить препаративные электрохимические методы получения нитроксильных радикалов из этих соединений. Показано, что промежуточным продуктом в синтезе нитроксильных радикалов из пространственно-затрудненных аминов ряда 2,2,6,6- тетраметилпиперидина является соответствующий 1-хлор- или 1-бром-2,2,6,6- тетраметилпиперидин.
3. Впервые показано, что электрохимическое окисление 1-хлор(бром)-производных ряда 2,2,6,6- тетраметилпиперидина является обратимым и приводит к образованию соответствующих устойчивых катион-радикалов, образование которых подтверждено методами ЦВА и ЭПР. На основе данных ЦВА, ЭПР и препаративного электролиза предложен механизм окисления 1-хлор-2,2,6,6- тетраметилпиперидина до НР и оксоаммониевого катиона.
4. Установлено, что электрохимическое поведение пространственно-затрудненных аминов ряда 2,2,6,6-тетраметилпиперидина отличается от поведения 1-галогенаминов ряда 2,2,6,6- тетраметилпиперидина. Показано, что процесс окисления пространственно-затрудненных аминов необратим и приводит к образованию неустойчивых катион-радикалов, которые превращаются в соответствующие аминильные радикалы, а затем нитроксильные радикалы. ЭПР-спектры этих радикалов зафиксированы при проведении электролиза в ячейке ЭПР-спектрометра.
5. Впервые проведено селективное электрохимическое окисление спиртов каталитической системой нитроксильный радикал ряда 2,2,6,6- тетраметилпиперидина – йодид калия до соответствующих карбонильных соединений. Первичные спирты в этом случае окисляются только до альдегидов. Процесс образования карбонильных соединений проходит с выходом 80-95 % по веществу и 40-47 %по току.
6. Показано, что в качестве катализатора в реакциях непрямого электрохимического окисления спиртов вместо НР можно использовать амины и 1-галогенпроизводные ряда 2,2,6,6-тетраметилпиперидина, при этом окисление спиртов проходит с хорошим выходом по веществу (65-95 %).
7. Установлено, что при электролизе раствора 4-оксо-2,2,6,6- тетраметилпиперидина (ТАА) в соляной кислоте основным продуктом реакции является 3,3,5,5-тетрахлор-4-оксо-2,2,6,6-тетраметилпиперидин. Изучено влияние на процесс температуры, плотности тока, материала анода, количества пропущенного электричества. Предложен способ получения 3,3,5,5-тетрахлор-4-оксо-2,2,6,6- тетраметилпиперидина с выходом по веществу 70-80 %.
8. Разработан способ получения гидробромида 3,5-дибром-4-оксо-2,2,6,6- тетраметилпиперидина (выход по веществу 80-85 %) электролизом раствора бромистоводородной кислоты или электролизом системы бромистый калий-серная кислота-вода.
9. Показано, что электрохимическое галогенирование 4-оксо-2,2,6,6- тетраметилпиперидина в кислой среде происходит путем взаимодействия образующегося в электрохимическом процессе хлора или брома с 4-оксо-2,2,6,6- тетраметилпиперидином.
10. Электрохимическое галогенирование 4-оксо-2,2,6,6-тетраметилпиперидина в щелочной среде в присутствии аммиака или в растворе метилата (этилата) натрия сопровождается перегруппировкой Фаворского и приводит к образованию смеси амидов или эфиров ряда тетраметилпирролина и тетраметилпирролидина. Разработаны препаративные электрохимические методы получения 3-карбоксамидо-2,2,5,5-тетраметилпирролидина, 3-карбметокси- и 3-карбэтокси-2,2.5,5-тетраметилпирролидина при восстановлении на ртутном катоде смеси соответствующих пирролинов и пирролидинов.
11. Разработаны новые методы синтеза пространственно-затрудненных аминов, моно- и бирадикалов ряда 2,2,6,6-тетраметилпиперидина и 2,2,5,5-тетраметилпирролидина на основе тетра- и дигалогенпроизводных 4-оксо-2,2,6,6-тетраметилпиперидина и научно-техническая документация на их производство.
Основное содержание диссертации опубликовано в следующих работах
(научные статьи, монографии, патенты):
1. Ожогина, О.А. Аномальная устойчивость дииминоксилпинакона в условиях дегидратации./ О.А. Ожогина, Е.Ш. Каган, И.Ю. Жукова, И.С. Кашпаров // Докл. АН СССР.- 1990.-Т.310, №3.- С. 619-621.
2. Каган, Е.Ш. Нитроксильные радикалы и пространственно-затрудненные амины, их применение/ Е.Ш. Каган, И.Ю. Жукова// Реактив: РИОР - 1992.-Вып.8.-С.12-13.
3. Каган, Е.Ш. Синтез и свойства 3,5-дибром-4-оксо-2,2,6,6-тетраметилпиперидин-1-оксила./ Е.Ш. Каган, И.Ю. Жукова, В.А. Смирнов //Химия гетероциклических соединений. -1992.-№ 1.-С. 73-74.
4. Жукова, И.Ю. Электрохимическое хлорирование и бромирование производных 2,2,6,6-тетраметилпиперидина /И.Ю. Жукова, Е.Ш.. Каган, С.А. Пожидаева, В.А. Смирнов //Журнал органической химии.-1993.-Т. 29,-вып.4.-С.751-757.
5. Каган, Е.Ш. Электрохимическое иодирование триацетонамина. Синтез 3-карбоксамидо-2,2,5,5-тетраметилпиперидина. /Е.Ш. Каган, И.Ю. Жукова, С.А. Пожидаева, Е.И. Коваленко// Электрохимия.-1996.-Т.32, № 1.-С. 100-104.
6. Каган, Е.Ш. Электрохимическое бромирование триацетонамина в электролите бромистый калий - серная кислота/ Е.Ш. Каган, И.Ю. Жукова, С.А. Пожидаева, И.С. Кашпаров // Электрохимия.- 1996.-Т.32, № 6.- С. 781-783.
7. Каган, Е.Ш. Необычное поведение нитроксильных радикалов в процессе их электрохимического окисления на платиновом аноде./ Е.Ш. Каган, С.А. Пожидаева, И.Ю. Жукова, И.С. Кашпаров// Журнал органической химии. -1997.-Т. 33. Вып. 9. -С. 1439-1440.
8. Каган, Е.Ш. Электрохимические превращения триацетонамина / Е.Ш. Каган, И.Ю. Жукова.//Электрохимия. – 2000. - Т.36, №2. -С. 224-232.
9. Кашпарова В.П., Жукова И.Ю., Каган Е.Ш. Синтез и применение N-галогенпроизводных соединений ряда пиперидина Юж.-Рос. гос. техн. ун-т.- Новочеркасск, 2000.- 17 с.- Деп. в ВИНИТИ 13.12.2000 № 3127-В00.- Аннот. БУ/ ВИНИТИ деп. науч. работы.- № 2.- б/о 136.
10. Кашпарова, В.П. Электрохимический синтез N-галогенпроизводных соединений ряда 2,2,6,6-тетраметилпиперидина./ В.П. Кашпарова, Е.Ш. Каган, И.Ю. Жукова // Изв. вузов. Сев.-Кавк. регион. Естеств. науки.-2001.- №2.- С. 44-45.
11. Томилов, А.П. Препаративная органическая электрохимия: Учеб. пособие. 2-е изд. перераб. и доп. /А.П. Томилов, Е.Ш. Каган, В.А. Смирнов, И.Ю. Жукова - Юж. Рос. гос. техн. ун-т – Новочеркасск: ЮРГТУ, 2002.- 153 с.
12. Каган, Е.Ш. Электрохимическое хлорирование триацетонамина/ Е.Ш. Каган, И.Ю. Жукова, С.Н. Кривошеева// Изв. вузов. Сев.-Кавк. регион. Естеств. науки. - 2001. - №3. - С. 61-63.
13. Кашпарова, В.П. Синтез соединений ряда 2,2,6,6-тетраметилпиперидина/ В.П. Кашпарова, Е.Ш. Каган, И.Ю. Жукова, И.С. Кашпаров//Журнал прикладной химии. –2002.- Т.75. - Вып.4.- С.682-683.
14. Кривошеева, С.Н., Электрохимический синтез 3-карбметокси-2,2,5,5-тетраметилпирролидина/ С.Н. Кривошеева, И.Ю. Жукова, Е.Ш. Каган// Журнал прикладной химии. –2002.- Т. 75, - вып. 9.- С.1566-1568.
15. Кашпарова, В.П. Электрохимическое галогенирование 4-гидроксимино-2,2,6,6-тетраметилпиперидина/ В.П. Кашпарова, И.Ю. Жукова, Е.Ш. Каган //Журнал прикладной химии –2004.- Т. 77, вып. 11.- С. 1808-1810.
16. Кашпарова, В.П. Применение 1-галогенпроизводных ряда 2,2,6,6-тетраметилпиперидина в качестве окислителей и галогенирующих агентов/В.П. Кашпарова, Е.Ш. Каган, И.Ю. Жукова, Е.П. Ивахненко //Журнал прикладной химии. –2004.- Т. 77, вып. 6.- С. 978-980.
17. Кривошеева, С.Н. Исследование стабильности 1-галогенпроизводных соединений ряда 2,2,6,6-тетраметилпиперидина /С.Н. Кривошеева, И.Ю. Жукова, Е.Ш. Каган, А.Ю. Доморовская //Изв. вузов. Сев.-Кавк. регион. Естеств. науки. - 2005. - № 2. - С. 68-70.
18. Кашпарова, В.П.,. Электрохимическое моделирование реакции фотолиза 1-хлор- и 1-бром-2,2,6,6-тетраметилпиперидина. / В.П.Кашпарова, Е.Ш. Каган, И.Ю. Жукова, Е.В. Власова, И.Б. Ильчибаева //Электрохимия. -2007.– Т. 43.- № 10.– С. 1-3.
19. Каган, Е.Ш. Исследование свойств нитроксильных радикалов методом циклической вольтамперометрии / Е.Ш. Каган, В.В. Янилкин, Н.А. Настапова, И.Ю. Жукова, В.П. Кашпарова, И.И. Кашпаров // Изв. вузов. Сев.-Кавк. регион. Естеств. науки. Спец. выпуск. «Проблемы электрохимии и экологии».- 2008.- С. 23-26.
20. Каган, Е.Ш. О механизме электрохимического окисления 1-хлор-2,2,6,6-тетраметилпиперидина / Е.Ш. Каган, В.В. Янилкин, Н.А. Настапова, В.И. Морозов, И.Ю. Жукова, В.П. Кашпарова, И.И. Кашпаров // Журнал общей химии. - 2009.- Т. 7.- Вып. 5.- С. 828 – 831.
21. Пат. 2302410 РИ МКИ5 С07D 211/94, С25В 3/02, Заявл. 25.04.06. Опубл. 10.07.07. «Изобретения. Полезные модели». Бюл. № 19 // Электрохимический способ получения 2,2,6,6-тетраметилпиперидин-1-оксила/ Каган Е.Ш. Жукова И.Ю., Кашпарова В.П., Домаровская А.Ю.
22. Пат. 2351693 РИ МКИ5 С25В 3/02, С07С 45/29, С07С 47/54, С07С 49/603; Заявл. 31.07.07. Опубл. 10.04.09. «Изобретения. Полезные модели». Бюл. № 10 // Способ окисления спиртов до карбонильных соединений / Кашпаров И.И., Кашпарова В.П., Жукова И.Ю., Каган Е.Ш.
23. Григорьев И.А., Иртегова И.Г., Каган Е.Ш., Жукова И.Ю. Электрохимия органических соединений в начале XXI века /Под ред. В.П. Гультяя, А.Г. Кривенко, А.П. Томилова// М.: Компания спутник +, 2008. С.315-377.
24. Жукова И.Ю., Козаченко П.Н. Синтез, свойства и применение галогенпроизводных триацетонамина и других соединений ряда 2,2,6,6-тетраметилпиперидина. Монография. Шахты ГОУ ВПО «ЮРГУЭС», 2009. 125 с.
25. Каган, Е.Ш. Окисление спиртов электрохимически генерируемым йодом в присутствии нитроксильных радикалов. /В.П. Кашпарова, Е.Ш. Каган, И.Ю. Жукова, И.И. Кашпаров//Журнал прикладной химии. – 2010.- Т.83, вып.4, - С.581-583.
Автор выражает глубокую благодарность научному консультанту профессору Кагану Е.Ш., сотрудникам Института органической и физической химии Казанского научного центра РАН д.х.н. Янилкину В.В., к.х.н. Морозову В.И., к.х.н. Настаповой Н.В., а также сотрудникам кафедр ХТВМС ОФКХ и ТЭП ЮРГТУ (НПИ) за систематическое обсуждение результатов исследований и критические замечания.