

Реакции 1,3,7-триазапиренов и солей 7-алкил-1,3,7-триазапирения с o-нуклеофильными реагентами
На правах рукописи
Тищенко
Олеся Александровна
Реакции 1,3,7-триазапиренов и солей
7-алкил-1,3,7-триазапирения с O-нуклеофильными
реагентами
02.00.03 – органическая химия
Автореферат
диссертации на соискание ученой степени
кандидата химических наук
Астрахань – 2011
Работа выполнена в Ставропольском государственном университете
| Научный руководитель: | доктор химических наук, профессор Боровлев Иван Васильевич | |
| Официальные оппоненты: | доктор химических наук, доцент Тырков Алексей Георгиевич доктор химических наук, профессор Гончаров Владимир Ильич | |
| Ведущая организация: | Кубанский государственный технологический университет |
Защита диссертационной работы состоится «14 » октября 2011 года в 1400 часов на заседании объединенного диссертационного совета по защите докторских и кандидатских диссертаций ДМ 307.001.04. при Астраханском государственном техническом университете (АГТУ) по адресу: 414025, г. Астрахань, ул. Татищева, 16, АГТУ, 2-ой учебный корпус, ауд. 201
С диссертацией можно ознакомиться в библиотеке АГТУ (ул. Татищева, 16, АГТУ, главный учебный корпус).
Автореферат разослан « » сентября 2011 г.
Ученый секретарь
диссертационного совета,
кандидат химических наук, доцент Шинкарь Е. В.
ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ
Актуальность проблемы. На фоне известных достижений химии азинов, азолов и их конденсированных аналогов свойства пери-аннелированных азагетероароматических систем остаются практически неизвестными. Между тем, особый способ сочленения карбо- и гетероколец может иметь следствием и особые свойства таких гетероциклов. К их числу относится и объект нашего исследования – 1,3,7-триазапирен (1a), а также его производные и, прежде всего, соли 7-алкил-1,3,7-триазапирения (2).

С практической точки зрения интерес к аза- и полиазапиренам обусловлен изменением их биологической активности по мере включения в пиреновое кольцо атомов азота. Так, если моноазапирены проявляют мутагенную и канцерогенную активность, то производные наиболее изученных 4,9- и 2,7-диазапиренов проявляют анальгетическую, противовирусную, антибактериальную, а также противораковую активность. Механизм подобного действия обычно связывают с их известной способностью выступать в качестве интеркаляторов. Полиазапирены оказались востребованы также и в ходе бурного развития супрамолекулярной химии.
Поскольку 1,3,7-триазапирен относится к -дефицитным гетероароматическим соединениям, нас интересовали, прежде всего, его реакции с нуклеофильными реагентами. В соответствии с современными представлениями [O.N. Chupakhin, V.N. Charushin, H. van der Plas. Nucleophilic Aromatic Substitution of Hydrogen. Academic Press, San Diego, 1994, 367 p.] нуклеофильное замещение в ряду электронодефицитных аренов и гетаренов реализуется как двухстадийный процесс присоединения – отщепления (схема 1). На первой стадии возможно образование двух промежуточных продуктов: X- или Н-аддуктов.
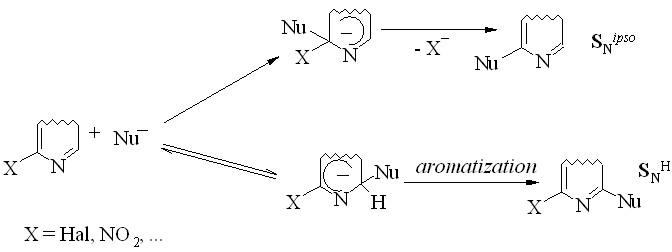
Схема 1
При этом присоединение нуклеофила к незамещенному атому углерода является, как правило, более быстрым процессом, чем ипсо-присоединение. Даже при наличии в кольце хорошего нуклеофуга ипсо-замещению предшествует быстрое и обратимое образование Н-аддуктов.
Ароматизация X-комплекса происходит путем необратимого элиминирования аниона Х-. Прямая же ароматизация Н-комплексов затруднена, поскольку предполагает отщепление гидрид-иона. Существует, однако, множество других путей трансформации Н-аддуктов в ароматические соединения, например, посредством элиминирования так называемых вспомогательных групп. В зависимости от расположения последних различают кине-замещение, теле-замещение, а также викариозное нуклеофильное замещение. Все эти методы имеют существенный недостаток: необходимость предварительного введения в молекулу субстрата или реагента вспомогательных групп.
Этого недостатка лишен метод прямого замещения атома водорода под действием нуклеофила (SNH). Поскольку отщепление гидрид-иона формально равносильно окислению субстрата, использование внешнего окислителя – наиболее очевидный способ ароматизации Н-комплекса. В настоящее время окислительное нуклеофильное замещение водорода (ONSH) активно изучается во всем мире.
Цель работы. Изначально целью работы было продолжение изучения реакции окислительного гидроксилирования солей 7-алкил-1,3,7-триазапирения. Результаты этой реакции, как было показано ранее в нашей лаборатории, существенно зависят от применяемого окислителя и условий её проведения. В ходе настоящей работы был сделан ряд неожиданных наблюдений. Самым интересным из них оказалось образование алкоксипроизводных самого 1,3,7-триазапирена. Изучение реакции SNH-алкоксилирования 1,3,7-триазапирена потребовало значительных усилий, поэтому раздел диссертации, связанный с синтезом и расщеплением простых эфиров данного гетероцикла, вышел на первый план, как по объему, так и по значимости. В конечном итоге нами была реализована и первоначальная цель работы, что позволило разработать удобный метод окислительного гидроксилирования не только солей 7-алкил-1,3,7-триазапирения, но и самого 1,3,7-триазапирена.
Научная новизна и практическая значимость. Впервые показано, что окислительное гидроксилирование солей 7-алкил-1,3,7-триазапирения легко протекает как в нейтральной среде, так и в условиях кислотного катализа с образованием 6-оксо-7-алкил-6,7-дигидро-1,3,7-триазапиренов с высоким выходом.
Найден метод синтеза 6-циано-8-оксо-7-алкил-7,8-дигидро-1,3,7-триазапиренов. Они являются продуктами двойного окислительного нуклеофильного замещения и образуются с хорошим выходом при взаимодействии солей 7-алкил-1,3,7-триазапирения с водным раствором NaCN и K3Fe(CN)6 при комнатной температуре.
При попытке гидроксилирования бромида 7-фенацил-1,3,7-триазапирения был получен продукт 1,3-диполярного циклоприсоединения, причем и 1,3-диполь, и диполярофил образуются из исходной соли. Исследование этой реакции позволило впервые получить серию производных новой гетеросистемы - пирроло[2.1-h][1,3,7]триазапирена.
Разработан метод окислительного SNH-алкоксилирования 1,3,7-триазапиренов первичными спиртами. Процесс протекает при комнатной температуре в системе вода/спирт/KOH/K3Fe(CN)6 и приводит сразу к продуктам двойного нуклеофильного замещения водорода – неизвестным ранее 6,8-диалкокси-1,3,7-триазапиренам.
Найдены условия, позволяющие, исходя из простых эфиров 1,3,7-триазапиренов, получать как продукты их моно-дезалкилирования – 6-оксо-8-алкокси-6,7-дигидро-1,3,7-триазапирены, так и продукты двойного дезалкилирования – 6,8-диоксо-1,6,7,8-тетрагидро-1,3,7-триазапирены.
Показано, что 1,3,7-триазапирены, в отличие от большинства азинов и азолов, легко подвергаются окислительному гидроксилированию в условиях кислотного катализа с образованием 6-оксо-6,7-дигидро-1,3,7-триазапиренов.
Исследованные реакции и разработанные методы синтеза отличаются простотой исполнения и могут найти применение в других классах -дефицитных гетероциклов для создания веществ с практически полезными характеристиками.
Апробация работы. Отдельные результаты работы докладывались на Международной конференции «Новые направления в химии гетероциклических соединений» (Кисловодск, 2009), Международной научной конференции «Фундаментальные и прикладные проблемы современной химии» (Астрахань, 2009), X Международном Семинаре по магнитному резонансу (Ростов-на-Дону, 2010), Международной конференции по химии гетероциклических соединений (Москва, 2010).
Публикации. Основное содержание работы опубликовано в 6 статьях и 6 тезисах докладов конференций.
Структура и объем диссертации. Диссертация состоит из введения, трех глав, выводов, списка литературы и приложения. Работа изложена на 106 страницах машинописного текста, иллюстрирована 10 таблицами и 8 рисунками. Библиография содержит 138 литературных ссылок.
В первой главе обобщены литературные данные по методам синтеза и свойствам четвертичных солей азапиренов. Вторая глава - обсуждение полученных нами результатов, причем раздел, посвященный алкоксилированию 1,3,7-триазапиренов, предваряется краткой сводкой литературных данных по SNH-алкоксилированию нитроаренов и гетаренов; третья глава – экспериментальная часть.
ОСНОВНОЕ СОДЕРЖАНИЕ РАБОТЫ[1]
1. Окислительное гидроксилирование солей 1-алкил-1,3,7-триазапирения
Для более детального изучения реакции окислительного гидроксилирования (реакция Деккера) мы, помимо уже известных солей 7-алкил-1,3,7-триазапирения, синтезировали ряд новых, включающих активную метиленовую группу. Как выяснилось, кватернизация 1,3,7-триазапирена (1a) аллилбромидом и бензилхлоридом, а также этилбромацетатом в сухом ацетонитриле завершается установлением равновесия, несмотря на большой избыток галогенида и длительное кипячение. Лучшие результаты были получены при кипячении реагентов в толуоле. В результате были впервые получены (схема 2) галогениды 7-аллил- (2f), 7-бензил- (2g) и 7-этоксикарбонилметил-1,3,7-триазапирения (2h). В отличие от почти бесцветного основания соли 2e-h представляют собой кристаллические вещества красно-коричневого цвета.
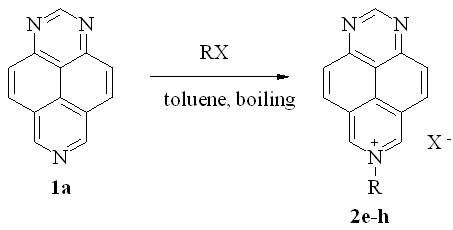
2: R = CH2COC6H5, X = Br (e); CH2-CH=CH2, X = Br (f);
CH2C6H5, X = Cl (g); CH2COOEt, X = Br (h)
Схема 2
1.1. Окислительное гидроксилирование в нейтральной среде
Ранее была обнаружена необычная способность солей 7-алкил-1,3,7-триазапирения (2a-c) подвергаться окислительному гидроксилированию даже в нейтральной среде. Реакция протекает при 75-80 оС в водном растворе окислителя (K3Fe(CN)6) в отсутствие щелочи с образованием 7-R-6-оксо-6,7-дигидро-1,3,7-триазапиренов (3a-c) (схема 3). Однако в случае солей 2a,b, помимо основных продуктов окислительного гидроксилирования 3a,b, из реакционной массы неожиданно были выделены с выходом 15-17% 6-циано-8-оксо-производные 4a,b, тогда как соль 2c образует только амид 3c (табл. 1, опыты 1-3).
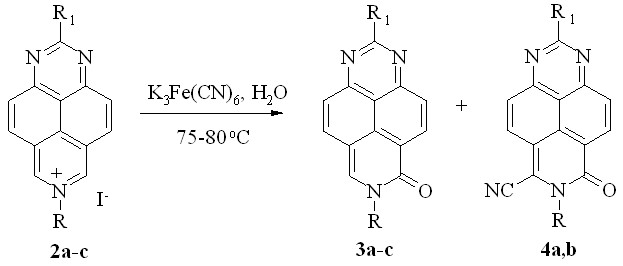
Схема 3
Продолжая изучение этой реакции, мы обнаружили, что цианопроизводные 4a,b не образуются в тех же условиях из амидов 3a,b. Это означает, что образование цианоамидов 4a,b начинается с окислительного нуклеофильного цианирования катионов 2a,b. С учетом того обстоятельства, что единственным источником цианид-иона в этих экспериментах является весьма прочный комплексный анион [Fe(CN)6]3-, можно сделать вывод, что содержащийся в очень малой концентрации, но гораздо более нуклеофильный, цианид-ион успешно конкурирует с молекулами воды в ходе присоединения к катиону субстрата. Таким образом, механизм образования соединений 4a,b включает в себя поэтапное присоединение к катионам 2a,b нуклеофилов с последующим окислением, причем на первой стадии присоединяется цианид-ион, а на второй – вода (схема 4).
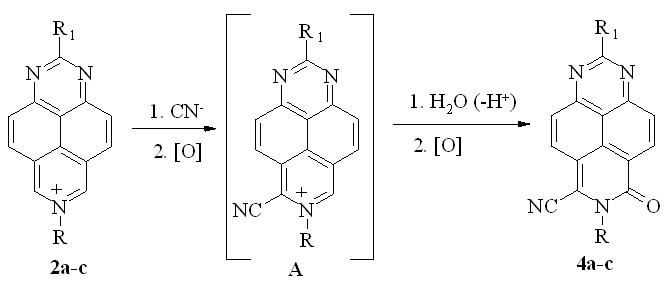
Схема 4
С учетом большей электрофильноcти промежуточно образующегося катиона А по сравнению с исходным катионом, вторая стадия протекает легко. Лимитирующим является, по-видимому, процесс окисления, поскольку на обеих стадиях ему подвергается нейтральная частица, а не анион, как в классическом исполнении реакции Деккера.
Цианоамиды 4a,b представляют значительный интерес, поскольку они являются результатом двойного ONSH-процесса, позволяющего вводить в субстрат сразу две разные функциональные группы. Естественно, на данном этапе целью работы была разработка препаративного метода синтеза соединений 4.
Логично было предположить, что увеличение концентрации цианид-аниона приведет к увеличению выхода цианоамидов 4. Действительно, взаимодействие не только солей 2a,b, но и соли 2с с избытком NaCN и K3Fe(CN)6 в водном растворе легко протекает при комнатной температуре (схема 5) с образованием 6-циано-8-оксо-7-алкил-7,8-дигидро-1,3,7-триазапиренов (4a-c) в качестве единственных продуктов реакции (табл. 1, опыты 4-6).
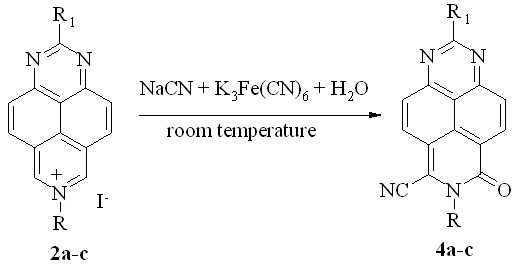
Схема 5
Как выяснилось далее, в измененных условиях, т. е. в системе NaCN/K3Fe(CN)6/H2O, соединения 3a-c также образуют цианоамиды 4a-c. По сути, это реакция окислительного нуклеофильного цианирования, и начинается она с нуклеофильного присоединения цианид-иона по азадиеноновому фрагменту молекул 3a-c с последующим окислением образующихся анионных интермедиатов (схема 6; табл. 1, опыты 7-9).
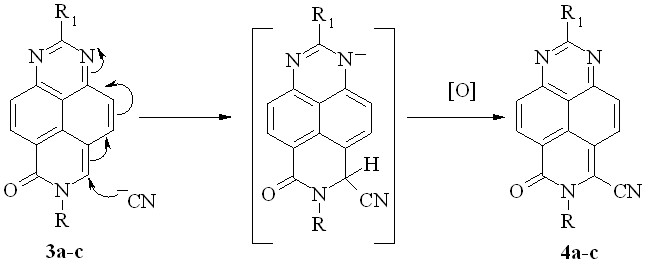
Схема 6
Таким образом, образование цианоамидов 4a-c из солей 7-алкил-1,3,7-триазапирения (2a-c) в системе NaCN/K3Fe(CN)6/H2O может протекать по обоим маршрутам: как цианирование - гидроксилирование, так и гидроксилирование - цианирование.
В ИК спектрах соединений 4a,b имеются как сигналы карбонильной группы (1657 и 1643 см-1), так и группы CN (2221 и 2223 см-1). В масс-спектре 4a наиболее интенсивным является пик молекулярного иона.
Таблица 1. Синтез 6-циано-8-оксо-7-алкил-7,8-дигидро-1,3,7-триазапиренов (4a-c) в водном растворе K3Fe(CN)6/H2O и в системе NaCN/K3Fe(CN)6/H2O
| Опыт | Исход. соедин. | R | R1 | Условия реакции | Продукт(ы) | |||
| 3 | Выход, % | 4 | Выход, % | |||||
| 1. | 2a | Me | H | K3Fe(CN)6/H2O, 75-80 oC | a | 73* [2] | a | 15* |
| 2. | 2b | Me | Me | K3Fe(CN)6/H2O, 75-80 oC | b | 70* | b | 17* |
| 3. | 2c | Et | H | K3Fe(CN)6/H2O, 75-80 oC | c | 69* | c | -* |
| 4. | 2a | Me | H | NaCN/K3Fe(CN)6/H2O, rt | - | - | a | 63 |
| 5. | 2b | Me | Me | NaCN/K3Fe(CN)6/H2O, rt | - | - | b | 61 |
| 6. | 2c | Et | H | NaCN/K3Fe(CN)6/H2O, rt | - | - | c | 54 |
| 7. | 3a | Me | Н | NaCN/K3Fe(CN)6/H2O, rt | - | - | a | 89 |
| 8. | 3b | Me | Me | NaCN/K3Fe(CN)6/H2O, rt | - | - | b | 83 |
| 9. | 3c | Et | H | NaCN/K3Fe(CN)6/H2O, rt | - | - | c | 75 |
Естественно, замена K3Fe(CN)6 на MnO2 в реакции солей 2a-c с водой приводит исключительно к 6-оксопроизводным 3a-c. Модернизируя этот подход, мы нашли далее, что удобным препаративным методом их синтеза оказалось кипячение этанольного раствора солей 2a-c в присутствии избытка MnO2: выход амидов 3a-c в этом случае близок к количественному (схема 7).
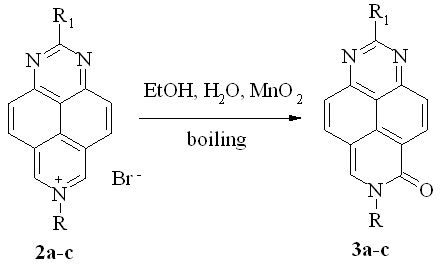
3: R1 = H, R = Me (a, 98%); R1 = R = Me (b, 97%); R1 = H, R = Et (c, 98%)
Схема 7
Однако попытка провести эту реакцию с солями 2e-g привела к смеси соответствующих продуктов окислительного гидроксилирования 3d-f и 1,3,7-триазапирена (1а; схема 8). Разделить смесь соответствующих амидов 3d-f и триазапирена (1а) нам не удалось вследствие близкой хроматографической подвижности и плохой растворимости этих соединений.
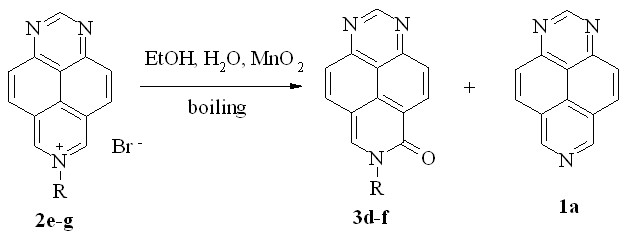
3: R = CH2COC6H5 (d); CH2-CH=CH2 (e); CH2C6H5 (f)
Схема 8
По данным ЯМР 1Н спектроскопии содержание в смеси продуктов гидроксилирования 3e,f составляет ~ 50%, а 3d ~ 35%. Это означает, что скорости SN2-процесса, приводящего к дезалкилированию солей 2e-g, и реакции их окислительного гидроксилирования сопоставимы, причем роль нуклеофильных агентов в обоих случаях выполняют молекулы воды.
1.2. Окислительное гидроксилирование в кислой среде
Известно, что процессу нуклеофильного замещения водорода способствует не только основная среда за счет образования более нуклеофильного, чем нейтральная частица, аниона, но и кислотный катализ (в более общем случае, электрофильный катализ).
Мы обнаружили, что при добавлении к раствору солей 2a-c в 60%-ной серной или в концентрированной соляной кислоте окислителя (K3Fe(CN)6) реакция легко протекает при комнатной температуре с образованием продуктов окислительного моно-гидроксилирования 3a-c (схема 9).
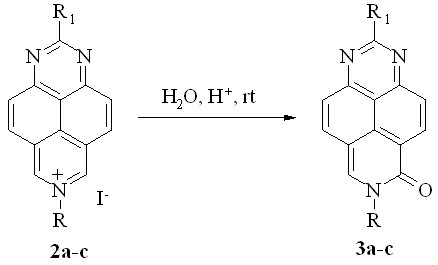
R1 = H, R = Me (a, 91%); R1 = R = Me (b, 86%); R1 = H, R = Et (c, 78%)
Схема 9
Вероятный механизм этого превращения (схема 10) включает протонирование катиона 2, нуклеофильную атаку образующегося дикатиона молекулой воды и последующее окисление продукта ковалентной гидратации – катиона псевдооснования или самого псевдооснования. Возможным окислителем является образующаяся в этих условиях кислота – H3Fe(CN)6.
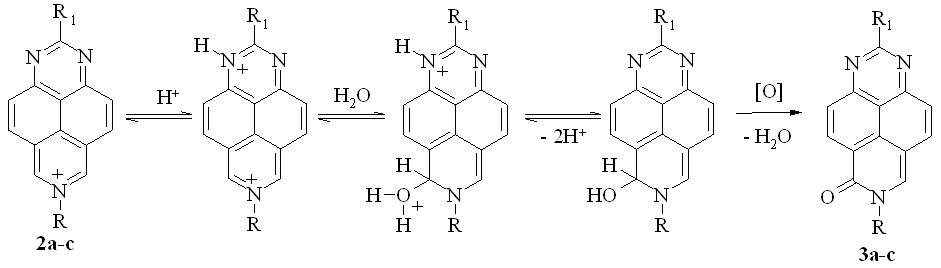
Схема 10
Попытки осуществить повторное гидроксилирование амидов 3a-c в более жестких условиях (повышение концентрации кислоты, увеличение времени реакции) не привели к успеху.
Таким образом, данный метод являет собой редкий пример проведения реакции Деккера в условиях кислотного катализа, позволяя в мягких условиях и с высоким выходом получать продукты окислительного моно-гидроксилирования солей 7-алкил-1,3,7-триазапирения.
1.3. Реакции солей 7-алкил-1,3,7-триазапирения в щелочной среде
Ранее было показано, что соли 2a-c, амиды 3a-c и цианоамиды 4a-c при нагревании с водной щелочью образуют продукты двойного окислительного гидроксилирования - 6,8-диоксо-7-алкил-1,6,7,8-тетрагидро-1,3,7-триазапирены (5a-c; схема 11).
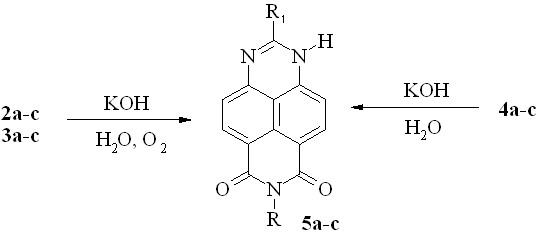
R1 = H, R = Me (a); R1 = R = Me (b); R1 = H, R = Et (c)
Схема 11
В роли окислителя при превращении солей 2 и амидов 3 выступает кислород воздуха (использование K3Fe(CN)6 приводит к окислительной деструкции соединений 2 и 3). Превращение цианоамидов 4a-c протекает как SNipso-замещение цианогруппы и не требует окислителя.
Однако при попытке провести подобную реакцию с бромидом 7-фенацил-1,3,7-триазапирения (2e) мы получили весьма неожиданный результат. При действии щелочи в водном или водно-диоксановом растворе на соль 2e при 75-80 оС вместо ожидаемых продуктов окислительного моно- или дигидроксилирования была получена смесь веществ, содержащая олигомеры красного цвета, 1,3,7-триазапирен (1а) и новый продукт с интенсивной оранжевой флюоресценцией в УФ свете (данные ТСХ). Из этой смеси мы выделили в чистом виде лишь обладающее наибольшей хроматографической подвижностью новое соединение. На основании комплекса спектральных и спектрометрических данных ему было приписано строение 10-бензоил-11-фенилпирроло[2.1-h][1,3,7]триазапирена (6а; схема 12).

Схема 12
Как выяснилось далее, это же соединение образуется и при взаимодействии соли 2e с триэтиламином в тетрагидрофуране.
Мы предположили, что соединение 6а образуется в результате 1,3-диполярного циклоприсоединения, причем в роли 1,3-диполя выступает илид 7, образующийся при депротонировании катиона 2e, а в роли диполярофила – енольная форма фенацильного производного 8, получающаяся in situ в результате SN2-реакции (схема 13). Ароматизация промежуточного аддукта 9 происходит путем отщепления молекул простых веществ (HX; H2O), т. е. окислитель в этой реакции не требуется.
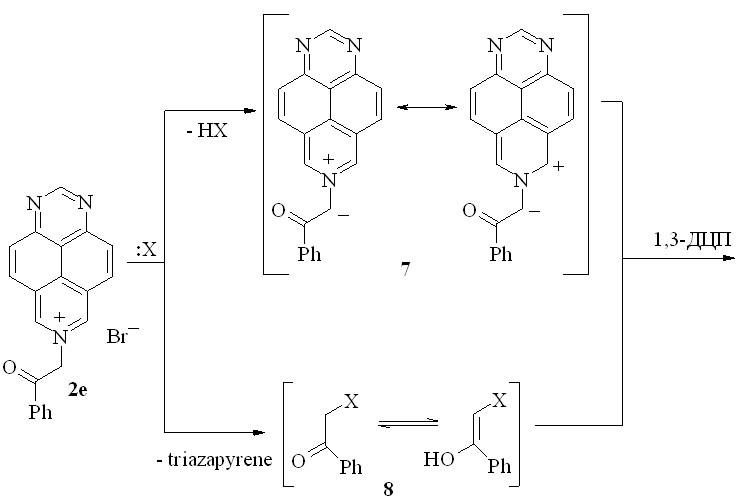
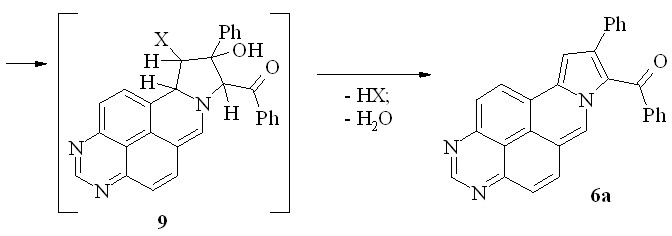
X = OH-, NEt3
Схема 13
Мы нашли далее, что, если в качестве диполярофила использовать соединения с активированной двойной связью, например, халконы 10, бромиды 7-фенацил-(2e) и 7-этоксикарбонилметил-1,3,7-триазапирения (2h) легко образуют продукты циклоприсоединения 11 красного цвета. Однако, ввиду их неустойчивости, реакцию проводили в присутствие окислителя (I2), получая неизвестные ранее производные пирроло[2.1-h][1,3,7]триазапирена (6b-g; схема 14 и табл. 2).

Схема 14
Таблица 2. Пирроло[2.1-h][1,3,7]триазапирены (6а-g)
| № | № соединения | R | R1 | R2 | Выход, % |
| 1. | 6a | Ph | Ph | H | 35 |
| 2. | 6b | Ph | Ph | Ph | 56 |
| 3. | 6c | Ph | Ph | 4-ClC6H4 | 52 |
| 4. | 6d | Ph | Ph | 4-O2NC6H4 | 42 |
| 5. | 6e | Ph | Ph | 4-Me2NC6H4 | 47 |
| 6. | 6f | Ph | Me | Ph | 50 |
| 7. | 6g | OEt | Ph | 4-ClC6H4 | 45 |
Попытки использовать для реакции другие растворители и окислители не привели к увеличению выхода соединений 6b-g. Не исключено, что окислительные свойства на стадии ароматизации отчасти проявляет и исходный катион 2; образующееся при этом неустойчивое дигидропроизводное 1,3,7-триазапирена вступает в процесс олигомеризации. Следует отметить также, что во всех случаях по данным ТСХ образуется небольшое количество 1,3,7-триазапирена и продукта «самосборки» 6а (из соли 2e).
При попытке провести реакцию соли 2e с циклогексеном мы получили лишь продукт 6а. Это позволяет сделать вывод о том, что выступающий в качестве диполярофила интермедиат 8 вступает в процесс циклоприсоединения с большей скоростью, чем неактивированный алкен.
Соединения 6а-g являются кристаллическими веществами; их окраска - от оранжевой до красно-коричневой. В УФ свете они характеризуются интенсивной флюоресценцией оранжевого цвета.
Характерной особенностью спектров ЯМР 1Н этих соединений являются слабопольные синглетные сигналы протонов в положениях 4 (10.5-10.7 м.д.) и 8 (9.6-9.7 м.д.).
В соответствии со строением в спектре ЯМР 13С соединения 6с имеется 30 сигналов, причем самые слабопольные пики ( 188.482 и 195.766 м. д.) относятся, несомненно, к атомам углерода двух карбонильных групп.
Весьма неожиданный результат мы получили при попытке окислительного гидроксилирования солей 7-фенацил- (2e), 7-аллил- (2f) и 7-бензил-1,3,7-триазапирения (2g) в водно-спиртовом растворе в присутствии щелочи и окислителя (K3Fe(CN)6). Реакция протекает неоднозначно, с образованием продуктов осмоления. Основным соединением, полученным из соли 2e, оказался, естественно, все тот же 10-бензоил-11-фенилпирроло[2.1-h][1,3,7]триазапирен (6а). Но главное то, что во всех трех случаях нами было выделено одно и то же новое соединение, которому на основании комплекса спектральных и спектрометрических данных было приписано строение 6,8-диэтокси-1,3,7-триазапирена (12a; схема 15).
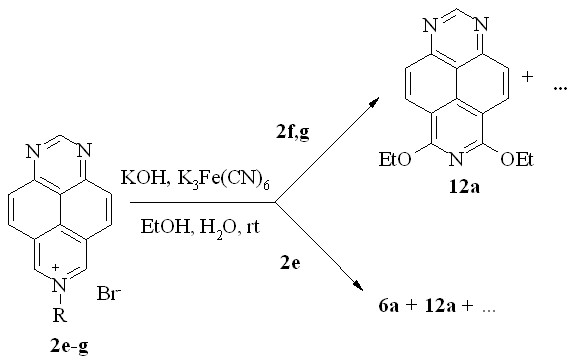
12a: 11% (из 2e); 36% (из 2f); 70% (из 2g)
Схема 15
Несложно было предположить, что образование соединения 12a является результатом SN2-дезалкилирования катионов 2e-g и последующего окислительного алкоксилирования 1,3,7-триазапирена. Это предположение подтвердилось, но с учетом важности и неизученности реакций SNH-алкоксилирования в ряду аренов и гетаренов рассмотрение полученных результатов мы вынесли в самостоятельный раздел 3.
2. Окислительное гидроксилирование 1,3,7-триазапиренов
Окислительное гидроксилирование азинов и азолов известно давно. Обычно его проводят сплавлением гетаренов с большим избытком безводной щелочи при высоких температурах. Однако, как выяснилось, в этих условиях 1,3,7-триазапирен (1a) полностью разрушается.
Исходя из результатов гидроксилирования солей 7-алкил-1,3,7-триазапирения (2a-c) в кислой среде, мы предположили возможность кислотного катализа и в случае протонных солей гетероцикла. Действительно, при действии на раствор 1,3,7-триазапиренов 1a,b в концентрированной соляной кислоте окислителем (K3Fe(CN)6) при комнатной температуре образуются продукты окислительного гидроксилирования - 6-оксо-6,7-дигидро-1,3,7-триазапирен (13a) и 6-оксо-2-метил-6,7-дигидро-1,3,7-триазапирен (13b) с выходом 71 и 50 %, соответственно (схема 16).
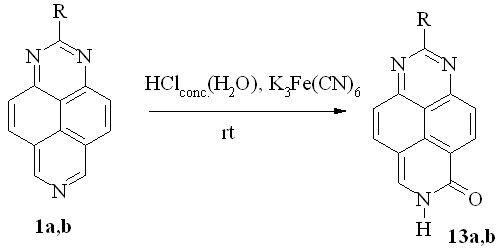
R = H (a), Me (b)
Схема 16
По аналогии с солями 7-алкил-1,3,7-триазапирения, вероятный механизм этого превращения включает двойное протонирование оснований 1a,b, нуклеофильное присоединение воды к дикатионам и последующее окисление (схема 17), причем вероятным окислителем является образующаяся в этих условиях кислота - H3Fe(CN)6.
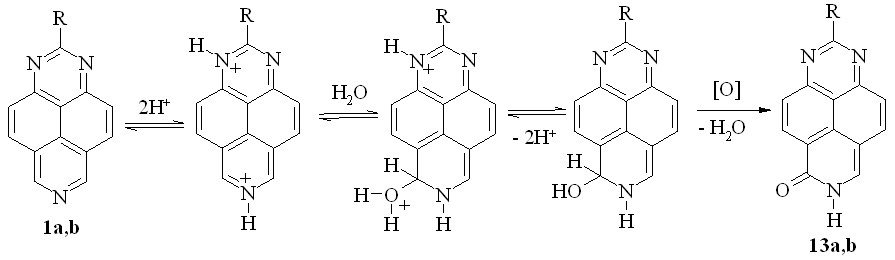
Схема 17
Соединения 13a,b в результате аннулярной прототропии могут существовать в четырех таутомерных формах. Однако сравнение спектров ЯМР 1H соединения 13a и его N7-метилированного аналога 3a позволило нам сделать однозначный вывод об их существовании в форме 6-оксо-6,7-дигидро-1,3,7-триазапирена.
ИК спектры кристаллических образцов соединения 13a,b также свидетельствуют в пользу амидного строения этих соединений.
Чтобы подтвердить возможность двойного протонирования 1,3,7-триазапирена, был снят спектр ЯМР 1Н осадка, полученного пропусканием сухого хлороводорода в толуольный раствор 1а (схема 18). Как следует из спектра полученного кристаллического продукта, он представляет собой смесь гидрохлорида (14) и дигидрохлорида (15) в соотношении ~ 9 : 2. С учетом вполне вероятной неустойчивости соли 77 можно с уверенностью предположить, что содержание дигидрохлорида в смеси было значительно выше. Впрочем, и без этих деталей понятно, что двойное протонирование 1,3,7-триазапирена протекает достаточно легко.
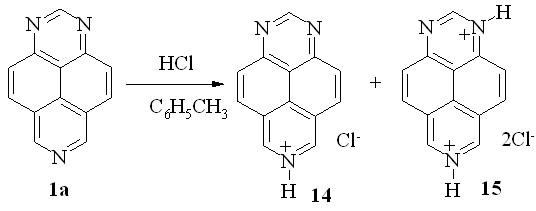
Схема 18
Данный метод окислительного гидроксилирования представляет интерес и с практической, и с теоретической точки зрения, поскольку являет собой редкий пример окислительного гидроксилирования гетероцикла в водной среде в условиях кислотного катализа. Он может найти применение в случае других -дефицитных гетероциклов.
3. Окислительное SNH-алкоксилирование 1,3,7-триазапиренов
В литературе описаны лишь единичные примеры успешного проведения реакций окислительного SNH-алкоксилирования. Тем удивительнее обнаруженная нами способность 1,3,7-триазапиренов вступать в реакцию нуклеофильного замещения водорода на алкоксигруппу в довольно необычных условиях. Она легко протекает при комнатной температуре при действии на триазапирены 1a,b избытком KOH и K3Fe(CN)6 в водно-спиртовом растворе. При использовании первичных спиртов - метилового, этилового, пропилового, аллилового и даже этиленгликоля – с высоким выходом получены неизвестные ранее продукты двойного алкоксилирования этого гетероцикла - 6,8-диалкокси-1,3,7-триазапирены (12a-f; схема 19, табл. 3).
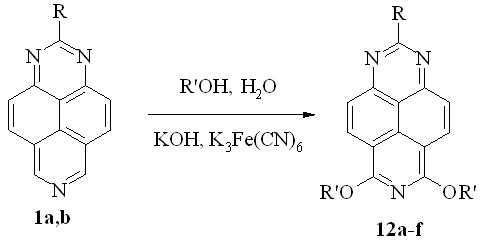
Схема 19
Таблица 3. Окислительное SNH-алкоксилирование 1,3,7-триазапиренов (1a,b)
| № | R | R’ | Время реакции, ч | Продукт | Выход, % |
| 1 | H | Et | 5 | 12a | 91 |
| 2 | H | Me | 2 | 12b | 98 |
| 3 | Me | Me | 3 | 12c | 89 |
| 4 | H | Pr | 16 | 12d | 69 |
| 5 | H | All | 6 | 12e | 36 |
| 6 | H | CH2CH2OH | 20 | 12f | 67 |
Попытки остановить реакцию на стадии моно-алкоксилирования успехом не увенчались: даже при недостатке окислителя образуются диалкоксипроизводные 12, и остается исходный триазапирен (1a). Это означает, что вторая стадия алкоксилирования протекает легче, чем первая.
Моно-метоксипроизводное 16a удалось получить (схема 20) лишь в случае 2-метил-6-фенил-1,3,7-триазапирена (1g), когда одно из -положений к N7-атому занято заместителем.
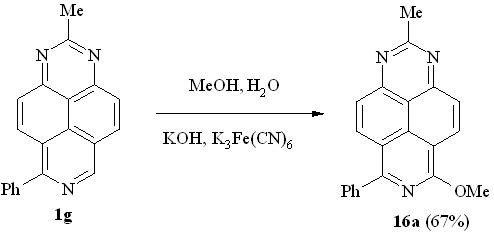
Схема 20
Если заняты оба -положения, как в случае 6,8-дифенил-1,3,7-триазапирена, реакция не протекает.
Необычность реакции алкоксилирования 1,3,7-триазапиренов заключается в том, что протекает она в исключительно мягких условиях, с высокой избирательностью субстрата в отношении имеющихся в реакционной системе нуклеофилов, а также в легкости повторного нуклеофильного замещения. Последовательное исключение отдельных компонентов реакционной смеси показало, что в отсутствие щелочи или K3Fe(CN)6 реакция не протекает, в отсутствие воды она, как минимум, очень сильно замедляется. Вероятно, вода необходима для увеличения растворимости окислителя, поскольку в спиртах он малорастворим.
С учетом этих факторов мы предположили следующий механизм этой реакции (схема 21):
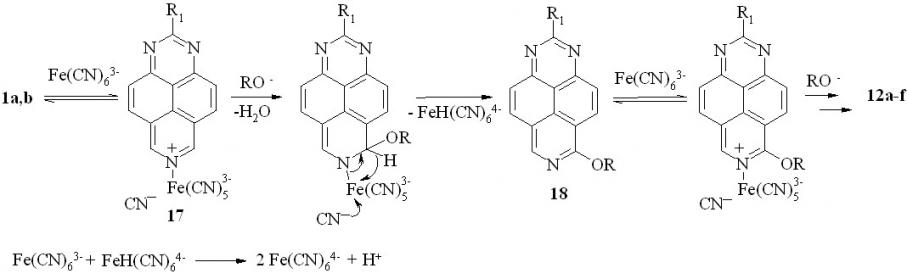 Схема 21
Схема 21
Он включает обратимое образование пентацианоферратного комплекса с 1,3,7-триазапиренами (17). Обладая весьма сильными акцепторными свойствами, этот заместитель генерирует высокий положительный заряд в -положениях триазапиренового цикла, что и провоцирует нуклеофильную атаку алкоксид-анионом. Далее под действием цианид-иона комплекс с гетероциклом разрушается с синхронным или постадийным акцептированием гидрид-иона окислителем (как это было показано ранее для реакции Деккера). Повторное алкоксилирование 18 протекает аналогично, причем, судя по результату, оно протекает легче, чем в случае исходного триазапирена. Таким образом, ферроцианид калия выполняет сначала функцию электрофильного катализатора реакции, а затем – окислителя.
Как выяснилось, пентацианоферратные комплексы таких гетероциклов, как пиридин, имидазол, бензимидазол и других, известны и используются в неорганической биохимии в качестве модельных соединений для изучения процесса переноса электрона в биологических объектах.
Использование в данной реакции вместо K3Fe(CN)6 других окислителей (KMnO4, MnO2, H2O2, нитробензол или натриевая соль м-нитробензолсульфокислоты) оказалось неэффективным. Селективность реакции в отношении присутствующих в системе нуклеофилов можно объяснить условиями кинетического контроля: реагирует более нуклеофильный алкоксид-анион, присутствующий в системе в небольшой равновесной концентрации.
Впрочем, с учетом известных примеров электроноакцепторных свойств алкокси-групп, находящихся в -положении к пиридиновому атому азота, нельзя отрицать и возможность прямой нуклеофильной атаки интермедиата 18 алкоксид-анионом с последующей дегидроароматизацией образующегося -аддукта.
При взаимодействии 1,3,7-триазапирена (1a) со вторичным спиртом (2-пропанол) наряду с 6,8-диизопропокси-1,3,7-триазапиреном (12g) образуется 6-изопропокси-1,3,7-триазапирен (16b, схема 22). Вследствие одинаковой хроматографической подвижности разделить эту смесь нам не удалось; по данным хроматомасс-спектрометрии и ЯМР 1H спектроскопии соотношение продуктов 12g и 16b составляет ~ 1 : 1. С увеличением времени реакции, однако, суммарный выход этих соединений снижается, и появляется уже известный продукт окислительного гидроксилирования 13а (данные ТСХ), по-видимому, в результате процесса E2-элиминирования изопропоксипроизводного 16b.
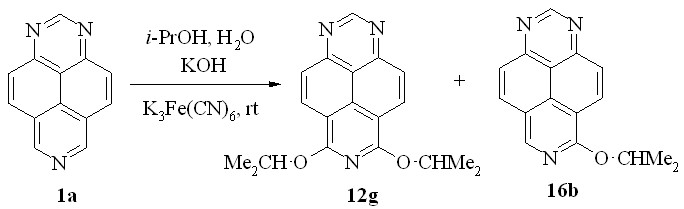
Схема 22
Попытка использовать трет-бутиловый спирт для алкоксилирования 1,3,7-триазапирена (1a) показала, что реакция требует значительно большего времени для ее проведения и приводит к продукту окислительного гидроксилирования - 6-оксо-6,7-дигидро-1,3,7-триазапирену (13a). Поскольку, как выяснилось, в отсутствие трет-бутанола, реакция не протекает, можно предположить, что промежуточно образующийся 6-трет-бутокси-1,3,7-триазапирен (Б) легко элиминирует изобутилен, образуя соединение 13a (схема 23).
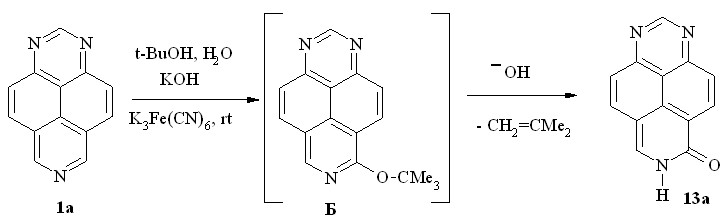
Схема 23
В спектрах ЯМР 1Н алкоксипроизводных 12a-f наиболее слабопольным, как и ожидалось, является протон в положении 2 ( 9.4 – 9.55); в области ароматических протонов спектры 12a-f весьма сходны и мало меняются при смене растворителя.
В масс-спектрах моно-метоксипроизводного 16а и диметоксипроизводных 12b,c самым интенсивным является пик молекулярного иона. Однако в случае диэтоксипроизводного (12a) наиболее интенсивным является пик [M - 56], дипропокси- (12d) и диизопропокси- (12g)
[3] – пик [M - 84], изопропокси- (16b)** – пик [M - 42], что соответствует отщеплению двух молекул соответствующего алкена (в последнем случае одной молекулы).
Мы нашли далее, что проведение реакции метоксилирования триазапирена (1a) в условиях кипячения реагентов (схема 24) несколько неожиданно привело к 8-метокси-6-оксо-6,7-дигидро-1,3,7-триазапирену (19).
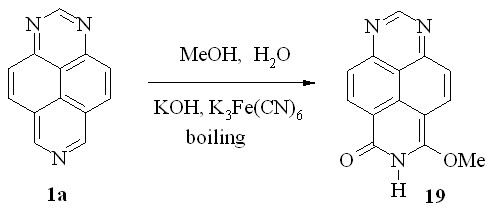
Схема 24
Формально можно представить несколько маршрутов образования соединения 19. Первый – окислительное гидроксилирование с последующим окислительным алкоксилированием – представляется маловероятным, поскольку продукт гидроксилирования в щелочной среде должен существовать в форме аниона, устойчивого к нуклеофилам. Второй путь – гидроксилирование промежуточного моно-метоксипроизводного – остается открытым, поскольку получить моно-метоксипроизводное пока не удалось.
Наиболее вероятным представляется третий путь образования соединения 19 – деметилирование промежуточного диметоксипроизводного 12b в результате SN2-реакции с гидроксид- или метоксид-анионом (схема 25).
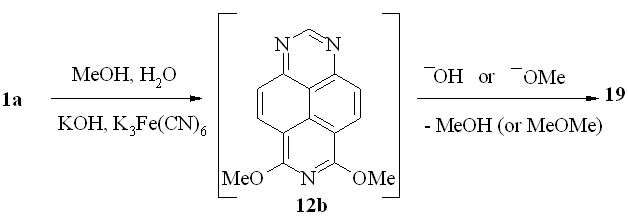
Схема 25
В специальном эксперименте мы показали, что при нагревании 6,8-диметокси-1,3,7-триазапирена (12b) с водной щелочью или метилатом натрия в метаноле он действительно подвергается моно-деметилированию с образованием соединения 19 в результате SN2-процесса (схема 25).
С другой стороны, давно известно, что простые эфиры азинов (в отличие от алкилариловых эфиров) легко подвергаются гидролитическому расщеплению при нагревании с разбавленными минеральными кислотами.
Мы нашли, что диалкоксипроизводные 12a,b при нагревании с 60%-ной серной кислотой подвергаются двойному гидролитическому расщеплению с образованием 6,8-диоксо-1,6,7,8-тетрагидро-1,3,7-триазапирена (20) (схема 26). По аналогии с известной работой [R. Daniels, L. T. Grady, L. Bauer. // J. Amer. Chem. Soc. –1965. –V. 87. –P. 1531], можно предположить, что и в данном случае реакция протекает в соответствии с SNipso-механизмом.
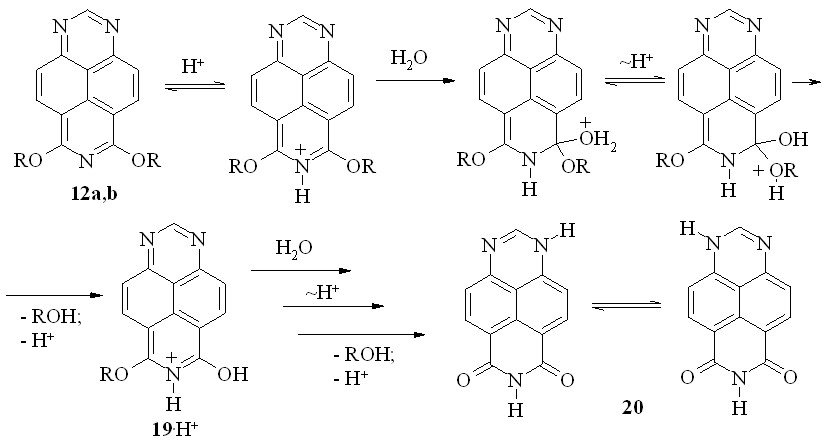
R = Et (a), Me (b)
Схема 26
Естественно, что и продукт щелочного моно-деметилирования – 8-метокси-6-оксо-6,7-дигидро-1,3,7-триазапирен (19) – в условиях кислотного катализа также образует имид 20 (схема 26). Особенностью спектра ЯМР 1H соединения 20 в DMSO-d6 является его симметризация вследствие быстрой в шкале времени ЯМР миграции протона между N1- и N3-атомами.
Таким образом, в зависимости от условий проведения реакции расщепления простых эфиров ряда 1,3,7-триазапирена можно получать как продукты их моно-дезалкилирования, так и продукты двойного дезалкилирования.
В целом же следует отметить, что особый способ сочленения карбо- и гетероколец в молекуле 1,3,7-триазапирена имеет следствием и его особые свойства. К числу последних следует отнести необычайную легкость OSNH-реакций, протекающих в водном растворе в нейтральной, щелочной и кислой среде, а также склонность к двойному нуклеофильному замещению.
Выводы
1. Обнаружена уникальная способность 1,3,7-триазапиренов легко вступать в реакцию окислительного SNH-алкоксилирования первичными спиртами. Реакция протекает при комнатной температуре в системе вода/спирт/KOH/K3Fe(CN)6 и приводит in one pot к продуктам двойного нуклеофильного замещения водорода – неизвестным ранее 6,8-диалкокси-1,3,7-триазапиренам. Моно-алкоксипроизводное образуется лишь в том случае, если одно из -положений к N7-атому занято заместителем.
2. Установлено, что 6,8-диалкокси-1,3,7-триазапирены при действии щелочных реагентов образуют продукты моно-дезалкилирования – 6-оксо-8-алкокси-6,7-дигидро-1,3,7-триазапирены, тогда как условия кислотного катализа приводят к продуктам двойного дезалкилирования – 6,8-диоксо-1,6,7,8-тетрагидро-1,3,7-триазапиренам.
3. Найдено, что 1,3,7-триазапирены, в отличие от большинства азинов и азолов, легко подвергаются окислительному гидроксилированию в условиях кислотного катализа с образованием 6-оксо-6,7-дигидро-1,3,7-триазапиренов.
4. Показано, что окислительное гидроксилирование солей 7-алкил-1,3,7-триазапирения легко протекает в нейтральной среде, а также в условиях кислотного катализа с образованием 6-оксо-7-алкил-6,7-дигидро-1,3,7-триазапиренов с высоким выходом.
5. Взаимодействие солей 7-алкил-1,3,7-триазапирения с водным раствором NaCN и K3Fe(CN)6 при комнатной температуре приводит к продуктам двойного окислительного нуклеофильного замещения – 6-циано-8-оксо-7-алкил-7,8-дигидро-1,3,7-триазапиренам. Те же соединения можно получить из 6-оксо-7-алкил-6,7-дигидро-1,3,7-триазапиренов, которые вступают в реакцию окислительного цианирования в тех же условиях.
6. Реакцией 1,3-диполярного циклоприсоединения в случае солей 7--оксоалкил-1,3,7-триазапирения впервые получены первые производные новой гетеросистемы – пирроло[2.1-h][1,3,7]триазапирена.
Основное содержание работы изложено в следующих публикациях:
1. О. П. Демидов, И. В. Боровлев, С. В. Писаренко, О. А. Немыкина (Тищенко). Необычная реакция солей 7–метил–1,3,7–триазапирения с водным раствором K3Fe(CN)6. // Химия гетероцикл. соедин. –2009. –№ 5. – С. 780-782.
2. И. В. Боровлев, О. П. Демидов, С. В. Писаренко, Н. В. Демидова, О. А. Немыкина (Тищенко). Соли 7–алкил–1,3,7–триазапирения: редкий случай окислительного гидроксилирования в условиях кислотного катализа. // Журн. орг. химии. –2009. –Т.45. –№ 11. –С. 1739-1740.
3. И. В. Боровлев, О. П. Демидов, С. В. Писаренко, О. А. Немыкина (Тищенко). Специфика реакций окислительного гидроксилирования солей 7–алкил–1,3,7–триазапирения. // Химия гетероцикл. соедин. –2010. – № 4. –С. 597-602.
4. О.П.Демидов, И.В.Боровлев, С.В.Писаренко, О.А.Немыкина (Тищенко). Соли 7–алкил–1,3,7–триазапирения: редкий случай двойного нуклеофильного замещения. // Журн. общей химии. –2010. –Т. 80. –№ 1. –С. 165-167.
5. О. П. Демидов, И. В. Боровлев, С. В. Писаренко, О. А. Немыкина (Тищенко), Н. А. Сайгакова. Окислительное SNH-алкоксилирование 1,3,7-триазапиренов. // Химия гетероцикл. соедин. –2010. –№ 5. –С. 791-793.
6. О. П. Демидов, И. В. Боровлев, Н. А. Сайгакова, О. А. Немыкина (Тищенко), Н. В. Демидова, С. В. Писаренко. Окислительное аминирование и гидроксилирование 1,3,7–триазапиренов в водной среде. // Химия гетероцикл. соедин. –2011. – № 1. –С. 142-144.
7. И. В. Боровлев, О. П. Демидов, С. В. Писаренко, О. А. Немыкина (Тищенко). Необычные аспекты окислительного гидроксилирования в ряду 1,3,7-триазапиренов и их солей. // Материалы Международной конференции «Новые направления в химии гетероциклических соединений». – Кисловодск. -2009. -С.46.
8. О. П. Демидов, И. В. Боровлев, С. В. Писаренко, О. А. Немыкина (Тищенко). Гидроксилирование 1,3,7-триазапиренов. // Материалы Международной конференции «Новые направления в химии гетероциклических соединений». – Кисловодск. -2009. -С.398.
9. О. А. Немыкина (Тищенко), С. В. Писаренко, О. П. Демидов, И. В. Боровлев. Окислительное гидроксилирование солей 7-алкил-1,3,7-триазапирения в кислой среде. // Материалы 3 Международной научной конференции «Фундаментальные и прикладные проблемы современной химии». – Астрахань: изд-во «Астраханский университет». -2009. -С. 55.
10. О. А. Немыкина (Тищенко), О. П. Демидов, И. В. Боровлев, С. В. Писаренко. Окислительное гидроксилирование солей 7-алкил-1,3,7-триазапирения. // Материалы X Международного Семинара по магнитному резонансу (спектроскопия, томография и экология). –Ростов-на-Дону. -2010. -С. 88.
11. И. В. Боровлев, О. П. Демидов, С. В. Писаренко, О. А. Немыкина (Тищенко), Н. А. Сайгакова. Нуклеофильное замещение в ряду 1,3,7-триазапиренов. // Международная конференция по химии гетероциклических соединений: Тезисы докладов, Москва. –2010. –С. 9.
12. О. П. Демидов, С. В. Писаренко, О. А. Немыкина (Тищенко), И. В. Боровлев, Н. А. Сайгакова. 1,3-Диполярное циклоприсоединение в ряду солей 7-алкил-1,3,7-триазапирения. // Международная конференция по химии гетероциклических соединений: Тезисы докладов, Москва. –2010. –С. 149.
Тищенко Олеся Александровна
Автореферат
диссертации на соискание ученой степени
кандидата химических наук
Подписано в печать. 31.08.2011 г.
Формат 60х84 1/16. Бумага офсетная.
Усл. печ. л. 1,34. Уч. изд. л. 0,98.
Заказ 564. Тираж 100 экз.
Отпечатано в Издательско-полиграфическом комплексе
Ставропольского государственного университета.
355009, г. Ставрополь, ул. Пушкина, 1.
[1] Автор выражает благодарность научному консультанту, к.х.н., доц. Демидову Олегу Петровичу
* Результаты, полученные ранее (Писаренко С.В., канд. диссертация, 2008).
Разделение смеси 12g и 16b и запись их масс-спектров проводилась с помощью хроматомасс-спектрометра.