

Анализ и прогноз биологической активности соединений на основе физико-химических закономерностей
На правах рукописи
ГРИШИНА МАРИЯ АЛЕКСАНДРОВНА
АНАЛИЗ И ПРОГНОЗ БИОЛОГИЧЕСКОЙ АКТИВНОСТИ СОЕДИНЕНИЙ НА ОСНОВЕ ФИЗИКО-ХИМИЧЕСКИХ ЗАКОНОМЕРНОСТЕЙ
02.00.04 – физическая химия
А в т о р е ф е р а т
диссертации на соискание ученой степени
доктора химических наук
Уфа, 2012
Работа выполнена на кафедре химии фармацевтического факультета Челябинской государственной медицинской академии.
Официальные оппоненты: доктор химических наук, профессор
Герчиков Анатолий Яковлевич
доктор химических наук
Авдин Вячеслав Викторович
доктор биологических наук, профессор
Поройков Владимир Васильевич
Ведущая организация: Государственное образовательное учреждение
высшего профессионального образования
«Тверской государственный университет»
Защита состоится “_15_” _марта_______ 2012 г. в 14 часов на заседании диссертационного совета Д 212.013.10 в ФГБОУ ВПО «Башкирский государственный университет», по адресу: 450074, г. Уфа, ул. Заки Валиди, 32, химический факультет, ауд. 305, e-mail: [email protected]
C диссертацией можно ознакомиться в библиотеке Башкирского государственного университета.
Ваш отзыв в одном экземпляре, заверенный гербовой печатью, просим направлять по адресу: 450074, г. Уфа, ул. Заки Валиди, 32, Башкирский государственный университет, химический факультет, ученому секретарю совета доктору хим. наук, профессору Прочухану Ю.А.
Автореферат разослан " " 2012 г.
УЧЕНЫЙ СЕКРЕТАРЬ
диссертационного совета
доктор хим. наук, проф. Ю.А. Прочухан
ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ
Актуальность работы. Одним из важнейших направлений исследований современной химии является создание эффективных лекарственных препаратов. На разработку одного лекарственного препарата в среднем требуется от 5 до 16 лет с затратами, часто превышающими 1 млрд. долларов с учетом препаратов, не достигших рынка. В связи с этим важным является создание теоретических (in silico) методов исследования механизмов действия лекарственных средств, прогноза активности, виртуального дизайна новых препаратов. Данные подходы позволяют определить количественные взаимосвязи физико-химических характеристик процессов взаимодействия лекарственных средств с биологическими системами от параметров строения молекул, их комплексов с биологическими рецепторами. Полученные закономерности могут быть использованы в прогнозе активности новых соединений, дизайне новых эффективных лекарственных средств, что позволяет существенно снизить стоимость лекарственных средств и время на их разработку. Существующие методы требуют значительных модификаций, поскольку недостаточный учет наиболее важных видов взаимодействия в комплексах лекарственных средств с рецептором, вероятности существования различных таутомерно-конформационных форм молекул, подстройки лекарственного средства к рецептору, мультистадийности процесса биологического действия приводят к низкой прогностической ценности получаемых количественных моделей для описания биологической активности.
Целью работы является компьютерное моделирование биологического действия соединений с учетом таутомерно-конформационного (ТК) состояния молекул, распределения электронной плотности, взаимодействия с биологическим рецептором, возможности метаболизма для прогноза новых перспективных лекарственных средств с учетом их метаболических свойств.
Научная новизна. В работе впервые осуществлено
-развитие теоретических подходов, позволяющих оценивать энергии ТК форм молекул, вероятность их существования с учетом влияния водной среды (комбинация алгоритмов MultiGen и ProK). Для этого предложено моделировать процессы таутомерных переходов, включающих стадии протонирования и депротонирования молекул, оценивать свободные энергии Гиббса данных процессов на основании предсказанных кислотно-основных свойств соединений;
- модифицированы 3D/4D QSAR подходы, полученная модификация реализована в 3D/4D QSAR алгоритме BiS/Multiconformational (BiS/MC), осуществляющем рассмотрение внешнего поля молекул, представленного в виде суммы кулоновского и ван-дер-ваальсового потенциалов; впервые наложение биологически активных соединений осуществлено с использованием процедуры самосогласованного поля, использованной ранее в методах квантовой химии;
- модифицированы сеточные 3D/4D QSAR подходы, полученная модификация реализована в 3D/4D QSAR алгоритме ConGO, впервые осуществляющем анализ схожести биологически активных соединений путем их наложения друг на друга до максимального совпадения электронной структуры, что позволило получить закономерности биологической активности от величин электронной плотности в узлах сетки, разбивающей результат наложения структур, осуществлять поиск фармакофорных и антифармакофорных фрагментов электронной структуры молекул с счетом их ТК состояния;
- впервые для оценки электронной плотности соединений использованы AlteQ функции, найденные с помощью данных прецизионного рентгеноструктурного анализа (РСА) и являющиеся в отличие от квантово-химических функций электронной плотности постоянными для каждого элемента Периодической Системы. Показано, что электронная плотность молекул, описанная с помощью AlteQ функций, также хорошо согласуется с данными прецизионного РСА;
- впервые предложена процедура ограниченного докинга соединений в полости рецептора, по качеству превышающая процедуру докинга с использованием существующих в мире аналогов, основанная на использовании данных РСА комплекса «рецептор-лиганд» хотя бы для одного соединения выборки и результата наложения структур, полученного алгоритмом BiS/MC.
- произведена модификация теоретических подходов для анализа строения комплексов «рецептор-лиганд», реализованная в подходе CoCon и позволяющая устанавливать активные центры рецептора и лиганда, определять количественные взаимосвязи величины БА с параметрами строения комплексов, структура которых была установлена методами докинга, в качестве параметров строения использованы энергетические (энергии водородных связей, ван-дер-ваальсовых и кулоновских взаимодействий) и силовые характеристики, рассчитанные с использованием непараметрических подходов.
- впервые совокупность этих методов была использована для анализа различных выборок биологически активных соединений - ингибиторов дигидрофолатредуктазы, p38 МАР киназы, нейраминидазы, ингибиторов 5-HT1A и 1-AR рецепторов. Кроме того, в работе рассмотрены субстраты 1А2, 2С9, 3А4 изоформ цитохрома р450, что позволяет оценивать возможность метаболизма соединений. Полученные закономерности позволили сделать прогноз новых перспективных лекарственных средств с учетом их метаболических свойств.
- с помощью предложенных в работе подходов и моделей найдены новые эффективные соединения противоопухолевого и противовоспалительного действия, прошедшие успешные предклинические испытания.
Практическая значимость. Разработанные методы дают возможность направленного создания новых перспективных лекарственных средств с учетом возможности их метаболизма в живых организмах.
Апробация. Основные положения диссертации доложены на 13th European Symposium on Quantitative Structure-Activity Relationships (Dusseldorf, Germany, 2000), 2-ой и 3-й Всероссийских конференциях “Молекулярное моделирование” (Москва, 2001, 2003), 33rd crystallographic course “From Genes to Drugs via Crystallography” (Erice, Italy 2002), I - V Национальной Конференции “Информационно-вычислительные технологии в решении фундаментальных научных проблем и прикладных задач химии, биологии, фармацевтики, медицины” (Москва, 2002, 2003, 2004, 2005, 2006), I Российской школе-конференции “Молекулярное моделирование в химии, биологии и медицине” (Саратов, 2002), XI - XIII Симпозиумах по межмолекулярному взаимодействию и конформациям молекул (Саратов, 2002; Санкт-Петербург, 2006, Челябинск, 2008), DPhg- Doktorandentagung (Germany, Dusseldorf, 2003), Fock School on computational and quantum chemistry (Novgorod the Great, Russia, 2003), III и IV Национальной кристаллохимической конференции (Черноголовка, 2003, 2006), IX and X International Seminar on Inclusion Compounds (Novosibirsk, Russia, 2003), 16th European Symposium on Quantitative Structure - Activity Relationships and Molecular Modelling (Mediterranean Sea, Italy, 2006), International Conference on Medicinal Chemistry (Istanbul, Turkey, 2007), Virtual Discovery-2007 and 2008 (London, Great Britain, 2007, Palm Springs, USA, 2008), MedChemEurope-2008 (Стокгольм, Швеция), “ADMET Europe-2010” (Мюнхен, Германия), International conference on medicinal chemistry (Брюссель, Бельгия, 2010), Green chemistry (Вашингтон, США, 2011)
Структура и объем диссертации. Диссертационная работа общим объемом 244 страницы состоит из введения, четырех глав, выводов, списка используемой литературы, включающего 290 наименований. Диссертация содержит 21 таблицу, 46 рисунков, 3 приложения.
ОСНОВНОЕ СОДЕРЖАНИЕ РАБОТЫ
Первая глава посвящена обзору существующих в мире компьютерных методов для анализа и прогноза биологической активности соединений, рассмотрены их преимущества и недостатки, определяющие низкую прогностическую ценность моделей.
Во второй главе приведены разработанные в рамках данной работы компьютерные методы моделирования биологической активности (БА) соединений, учитывающие трехмерную структуру молекулы, которая представляет собой набор ядер, окруженных электронной плотностью. Такая совокупность ядер, связанных друг с другом химической связью, рассматривается как прообраз скелета, изменение геометрии которого осуществляется за счет таутомерных и/или конформационных превращений, возникающих в результате подстройки под окружение, например, при взаимодействии с рецептором, растворителем и др. На рис. 1 представлено значительное изменение геометрии молекулы в зависимости от условий на примере конформеров ингибитора риновируса HRV14.
Для решения первой задачи – определения ТК формы молекулы в момент взаимодействия с рецептором –использована комбинация алгоритмов BiS и MultiGen, названная BiS/MC. Алгоритм MultiGen позволяет определить конформеры для каждого из предполагаемых таутомеров. Алгоритм BiS/MC рассматривает мультитаутомерно-конформационную модель биологически активных соединений, выделяет конформеры, ответственные за связывание молекулы лекарственного средства с рецептором. Для решения второй задачи – оценки электронной плотности молекул –использованы методы квантовой химии или альтернативные подходы.
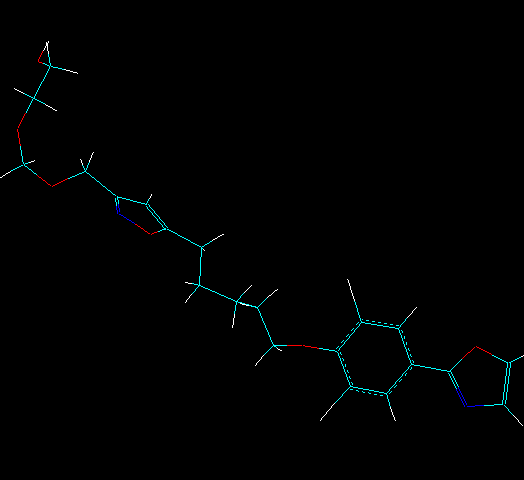
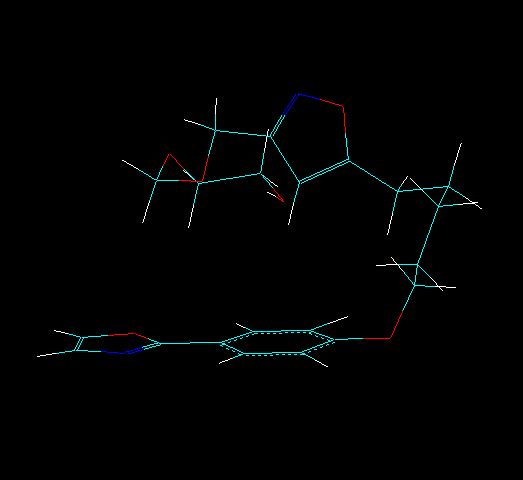
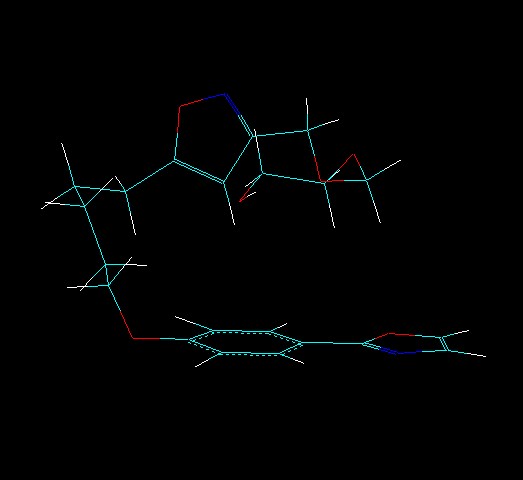

Рис. 1. Ингибитор HRV14: а) при взаимодействии с рецептором, б) и в) – изоэнергетические зеркально противоположные конформеры (энантиоконформеры) в газовой фазе.
В п.п. 2.1 рассмотрено построение мультитаутомерно-конформационной модели молекул с использованием алгоритма MultiGen. В его основе лежит предположение о том, что любые конформационные изменения в молекуле должны осуществляться вдоль направлений движения атомов во время колебаний. Поэтому для определения данных направлений необходимым является решение прямой спектральной задачи, определение мод колебаний. Тогда смещение координат атомов молекулы вдоль мод колебаний должно приводить к генерации новых конформеров. Поскольку каждая ахиральная структура может существовать в виде двух изоэнергетических зеркально противоположных конформеров (энантиоконформеров), форма которых также может иным образом определять возможность и эффективность взаимодействия с хиральным рецептором, в процедуре поиска конформеров осуществляется построение зеркально противоположной структуры путем изменения знака координаты атомов в найденном конформере на противоположный вдоль одной из трех координатных осей. Пример подобных энантиоконформеров представлен на Рис. 1б и в.
В случае существования соединения в различных таутомерных формах подобный мультиконформационный анализ проведен для каждого из таутомеров соединения. Тогда вероятность существования каждой ТК формы определяется по соотношению (1), а свойство (А) соединения представлено суперпозицией свойств (Ajl) всех ТК форм молекул (уравнение (2)).
![]() (1)
(1) ![]() (2)
(2)
где i и j - номера таутомеров, k – номер рассматриваемого конформера для таутомера под номером i; J – общее количество таутомеров; l – номер конформера для таутомера под номером j; Lj – общее количество конформеров в пределах 3 ккал/моль по отношению к наиболее выгодному конформеру таутомера j, Eik и Ejl – полные энергии ТК форм.
Для определения энергий ТК могут быть использованы квантовохимические методы с учетом влияния растворителя (воды), однако данные подходы требуют значительных временных затрат, что в подавляющем большинстве случаев делает их применение чрезвычайно сложным для решения подобных задач. Поэтому в данной работе предложен новый метод оценки энергий ТК форм с учетом влияния водного окружения, определяющего среду в биосистемах. Метод основан на рассмотрении процесса протолитической диссоциации соединения с отщеплением протона:
![]()
Данный процесс характеризует величина константы кислотности соединения АН - Ka. Тогда изменение свободной энергии Гиббса G процесса, характеризующего стабильность ТК формы, можно определить по уравнению Вант-Гоффа: ![]() .
.
Полная энергия ТК формы в водной среде с учетом энергий напряжения структуры может быть рассчитана по соотношению (3), причем меньшее значение полной энергии конформера с для таутомера t (Etc) соответствует большей стабильности структуры:
![]() (3)
(3)
Для расчета величин Ka предложено использовать алгоритм ProK. Данный алгоритм основан на моделировании взаимодействия структуры с пробным зарядом (протоном), помещенным на поверхность пространство-заполняющей модели молекулы, полученной с использованием силового поля MERA. Энергия электростатического взаимодействия пробного заряда, помещенного на поверхность атома m, с данным атомом m равна ![]() (где rm – ван-дер-ваальсов радиус атома m), с соседним атомом (i-ым атомом) молекулы эта величина составляет
(где rm – ван-дер-ваальсов радиус атома m), с соседним атомом (i-ым атомом) молекулы эта величина составляет ![]() (где Rmi – расстояние между атомами m и i). Полная энергия взаимодействия должна учитывать возможность собственной ионизации атома m, что было сделано путем расчета величины
(где Rmi – расстояние между атомами m и i). Полная энергия взаимодействия должна учитывать возможность собственной ионизации атома m, что было сделано путем расчета величины ![]() (где a0 Боровский радиус, IH – потенциал ионизации атома водорода), которая коррелирует с электроотрицательностью атомов. При расчетах был использован принцип полного выравнивания электроотрицательности [Mortier W.J., Van Genechten K., Gasteiger J. // J. Am. Chem. Soc.- 1985.- V. 107.- P. 829 - 835]. Выровненная электроотрицательность (n) атома, взаимодействующего с пробным зарядом, рассчитана с использованием системы уравнений (4):
(где a0 Боровский радиус, IH – потенциал ионизации атома водорода), которая коррелирует с электроотрицательностью атомов. При расчетах был использован принцип полного выравнивания электроотрицательности [Mortier W.J., Van Genechten K., Gasteiger J. // J. Am. Chem. Soc.- 1985.- V. 107.- P. 829 - 835]. Выровненная электроотрицательность (n) атома, взаимодействующего с пробным зарядом, рассчитана с использованием системы уравнений (4):
 (4) (4) | где - молекулярная характеристика, постоянная для всех атомов молекулы, а Q – заряд рассматриваемой структуры, в случае молекулы равный 0. Найдено, что величина связана с величинами pKа, измеренными в водных растворах при ионной силе, равной 0, и температуре 298 K, согласно соотношению: |
где a и b параметры регрессии. Коэффициент корреляции данной зависимости для выборки из 150 веществ, включающей амины, гетероциклы, карбоновые кислоты и т.д., составляет R=0.903 (коэффициент Фишера F=277).
Пример мультитаутомерно-конформационной модели молекулы представлен на Рис. 2. Всего для таутомера А найдено 5 конформеров, для таутомера Б найдено 7 конформеров. Таким образом, данная молекула может быть представлена 12 ТК структурами. Произведенная оценка вероятности существования каждой ТК формы согласно выражению (2) позволила определить суммарную вероятность существования таутомеров А и Б, которая составила 0, 585 и 0, 415 для таутомера А и Б соответственно.
 а) а)  б) б) |  в) в) |
Рис. 2. Мультитаутомерно-конформационная модель молекулы: а) и б) – наиболее выгодные таутомеры (соответственно А и Б) соединения по данным расчетов по формуле (3), в) результат наложения конформеров этих двух таутомеров с использованием алгоритма BiS/MC
Для подтверждения возможности использования полученной мультитаутомерно-конформационной модели соединений в п.п. 2.1 рассмотрены примеры сопоставления различных экспериментальных физико-химических характеристик соединений с расчетными, определенными в мультитаутомерно-конформационном приближении. Например, показано, что расчетная плотность 137 соединений выборки, для которых было найдено 1408 конформеров, коррелирует с экспериментальной (R = 0.9978, F = 30400; стандартное отклонение = 0.042).
Для анализа БА в п.п. 2.1 используется алгоритм BiS/MC, в основе которого лежит принцип комплементарности соединения полости рецептора. Ориентацию молекулы определяет весь комплекс ван-дер-ваальсовых, кулоновских и специфических взаимодействий с рецептором. В совокупности данные виды взаимодействий определяют молекулярное поле, которое и должно обеспечивать максимальную комплементарность к рецептору биологически активных соединений. При этом каждая молекула с данным видом БА содержит фрагменты, определяющие связывание с активными центрами рецептора. Эту часть молекулы можно назвать фармакофорной. Величина БА в значительной степени будет определяться эффективностью связывания фармакофорной части молекулы с рецептором. Остальные атомы, которые не взаимодействуют с рецептором, могут играть роль “балластной” части молекулы. Разные молекулы могут связываться с разными активными центрами рецептора. Поэтому генеральная совокупность всех активных молекул должна достоверно описывать поле рецептора. Тогда для решения проблемы ориентации молекул в полости рецептора необходимо определение совокупного поля выборки молекул.
Определение молекулярного поля в алгоритме BiS/MC проводится с использованием кулоновского потенциала и потенциала ван-дер-ваальсовых взаимодействий, наводимых на точку m поверхности молекулы. Кулоновский потенциал определяется из известного уравнения (5). Для расчета потенциала ван-дер-ваальсовых взаимодействий, наводимых на точку m, по аналогии с кулоновским использованы уравнения (6):
![]() (5)
(5) ![]() (6)
(6)
где N- число атомов рассматриваемой структуры, qi – заряд ее атома i, Rim – расстояние от точки m до атома i, k – коэффициент пересчета в единицы СИ, Vim- глубина минимума потенциальной энергии в соответствии с потенциалом Леннард-Джонса, ri – ван-дер-ваальсовый радиус атома i. Расчет величин Vim, ri и qi произведен в рамках модели MERA [Потемкин В.А., Барташевич Е.В., Белик А.В. // Журн. общ. химии.- 1995.- Т. 65.- № 2.- С. 205 - 208]. В уравнении (6) для упрощения расчетов учтено только притяжение за счет ван-дер-ваальсовых взаимодействий.
Комплементарное поле рецептора смоделировано в виде совокупности псевдо-атомов (пробных сфер с некоторым зарядом и радиусом), контактирующих с поверхностью молекулы. Для определения характеристик комплементарного поля вычисляются кулоновский и ван-дер-ваальсов потенциалы поля первой молекулы выборки. Полученные потенциалы позволяют определить по уравнениям (7) и (8) характеристики псевдо-атома с центром в точке m (заряд qm и радиус rm), который обеспечивает максимальную комплементарность рассматриваемой молекуле в данной точке поля. Набор псевдо-атомов является моделью рецептора.
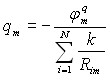 (7);
(7); 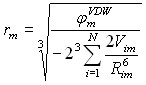 (8)
(8)
Процедура определения комплементарного поля, расчета характеристик псевдо-атомов производится для первой ТК формы молекулы, затем в комплементарном поле в рамках комбинированного симплексного и квази-ньютоновского метода проводится оптимизация ориентации второй ТК формы до достижения минимума совокупной вероятности контакта (P) ее атомов со всеми псевдо-атомами.
![]() , где
, где ![]() , М – число пробных сфер
, М – число пробных сфер
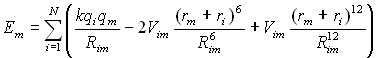 (9)
(9)
В найденной ориентации комплементарное поле рецептора уточняется путем добавки потенциалов поля второй ТК формы к имеющимся: ![]() ;
; ![]() . Потенциалы поля второй ТК формы
. Потенциалы поля второй ТК формы ![]() и
и ![]() вычисляются аналогично потенциалам первой формы по формулам (5) и (6). Аналогичная процедура осуществляется для третьей, четвертой и всех последующих ТК форм всех соединений выборки. Процедура прекращается, если различие в координатах атомов структур на новом и предыдущем шаге ориентации не превышает заданного предела. По окончании процедуры ориентации строится линейная зависимость между величиной БА и параметрами взаимодействий псевдо-атомов модельного рецептора с ТК формами.
вычисляются аналогично потенциалам первой формы по формулам (5) и (6). Аналогичная процедура осуществляется для третьей, четвертой и всех последующих ТК форм всех соединений выборки. Процедура прекращается, если различие в координатах атомов структур на новом и предыдущем шаге ориентации не превышает заданного предела. По окончании процедуры ориентации строится линейная зависимость между величиной БА и параметрами взаимодействий псевдо-атомов модельного рецептора с ТК формами.
![]()
где ![]() и
и ![]() - энергия и сила взаимодействия ТК формы cl (где с – номер конформера у татомера l) с активными центрами модельного рецептора,
- энергия и сила взаимодействия ТК формы cl (где с – номер конформера у татомера l) с активными центрами модельного рецептора, ![]() - вероятность существования ТК формы cl.
- вероятность существования ТК формы cl.
В качестве параметров использованы энергии взаимодействия, рассчитанные по уравнению (9), и силы![]() (10).
(10).
Для построения модели использованы регрессионные методы анализа, которые позволяют выделить те псевдоатомы, характеристики взаимодействия молекул с которыми описывают величину БА соединений с наибольшим коэффициентом корреляции. Данные атомы модельного рецептора представляют собой активные центры, а другие атомы модельного рецептора вносят менее существенный вклад в проявление биологического действия соединением, либо могут быть вариабельными вследствие конформационной подвижности рецептора.
По окончании данной процедуры определялась активность ТК формы cl (где c – номер конформера у таутомера l) по формуле:
![]()
Это позволяет выделить наиболее активные ТК формы соединения, которые в наибольшей степени определяют связывание молекулы с рецептором.
Возможность использования алгоритма BiS/MC для анализа БА проверена на значительном количестве выборок соединений, обладающих противотуберкулезным, антибактериальным, противовоспалительным, противоопухолевым эффектом, а также субстраты цитохрома Р450. При этом рассмотрены соединения различного строения и состава, включающие как типичные органогены, так и элементы высших периодов, в том числе переходные металлы. С использованием алгоритма BiS/MC определены фармакофорные части молекул, выявлены взаимосвязи БА с характеристиками взаимодействия в системах «модельный рецептор - лиганд» (выражения (5), (6), (9), (10)). Результаты данных исследований подробно представлены в главе 3. Показано, что коэффициенты корреляции полученных количественных моделей для описания величины БА составляют 0,90 – 0,99.
Для оценки возможности использования алгоритма BiS/MC в определении геометрии молекулы в момент ее связывания с рецептором проведено сопоставление расчетных наиболее активных ТК форм в случае соединений, имеющих несколько таутомеров, и наиболее активных конформеров в случае соединений, не претерпевающих таутомерных переходов, с данными РСА соответствующих комплексов «рецептор-лиганд», найденных в Protein Data Bank [Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. // Nucleic Acids Research.- 2000.- V.28.-p. 235-242]. На рис. 3а представлен пример подобного сопоставления – противоопухолевый ингибитор дигидрофолатредуктазы 1hfp[1] . Для данного соединения найдено 2910 ТК форм, представляющих собой набор более 50 таутомеров.
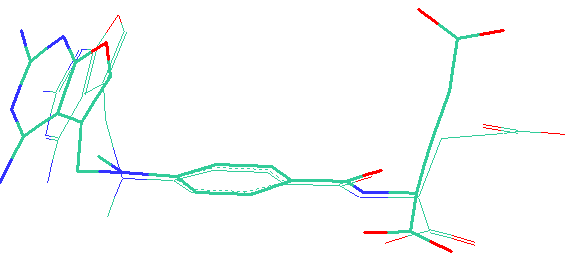
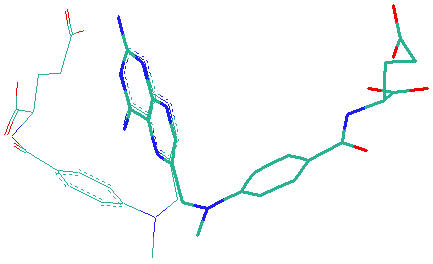
Рис. 3. Сравнение расчетных ТК форм ингибитора 1hfp со структурой, найденной РСА (выделена жирным, pGI50% (эксп.) =8,2): а) наиболее активной (pGI50% (расч.) =21,70); б) наименее активной (pGI50% (расч.) = –9,08).
На Рис. 3б показано сравнение наименее активной ТК формы данного соединения со структурой найденной РСА в комплексе. Данный рисунок демонстрирует значительные отличия между ними, что объясняет низкую расчетную БА для данной ТК формы соединения.
Конформационный анализ для противоопухолевого ДНК-антиметаболита 1d35 позволил выявить 39 конформеров, из которых с помощью алгоритма BiS/MC была выделена наиболее активная форма, представленная на Рис.4 в сравнении с данными РСА.
Использование алгоритма BiS/MC в мультиконформационном и мультитаутомерно-конформационном режиме позволяет определить строение «скелета» молекулы, взаимодействующей с рецептором. Показано, что расчетная геометрия молекулы, взаимодействующей с рецептором, хорошо согласуется с данными РСА комплексов «рецептор-лиганд». Для детального описания распределения электронной плотности в молекуле, связывающейся с рецептором, было предложено использовать алгоритм ConGO.
 |  |  |
Рис. 4. ДНК-антиметаболит 1d35: a) и б) результаты сравнения экспериментальной (выделена жирным) и расчетной ТК формы; в) 1d35 в комплексе с ДНК (определен РСА)
Данный алгоритм описан в п.п. 2.2.. В алгоритме ConGO рассматрены значения функции электронной плотности в узлах кубической сетки, разбивающей молекулу, с расстоянием между узлами, равным d (Рис. 5). В данном алгоритме молекулы выборки поочередно накладываются друг на друга путем операций вращения и трансляции до достижения максимального сходства в значении электронной плотности в узлах сетки. При этом критерием максимального сходства электронной плотности является минимальное значение величины:
![]() ,
,
где i – номер рассматриваемой молекулы,
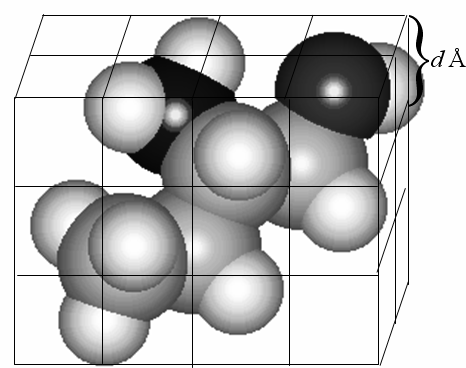 Рис. 5. Разбиение молекулы кубической сеткой с расстоянием между ближайшими узлами, равным d (обычно d=3 и менее). Рис. 5. Разбиение молекулы кубической сеткой с расстоянием между ближайшими узлами, равным d (обычно d=3 и менее). |
После этого весь цикл процедуры наложения структур повторяется заново для уточнения результата наложения. Итеративная процедура завершается, если различие электронных плотностей в узлах сетки на текущем и предыдущем шагах не превышает 0.1. После этого производится уточнение ориентации молекул друг относительно друга с разбиением структур сеткой с шагом 0,1d. Определенная ориентация соответствует максимальному совпадению наиболее схожих фрагментов электронной структуры молекул. Далее строится зависимость БА соединений от значений электронной плотности в узлах сетки с использованием методов регрессионного анализа. Уравнение имеет вид:
![]()
где t - номер узла сетки, значение в котором электронной плотности приводит к повышению величины БА, u – номер узла сетки, значение электронной плотности в котором приводит к понижению величины БА, с1, с2, с3 – параметры.
Полученная модель является двухфакторной, одним из факторов является сумма электронных плотностей в узлах сетки, приводящих к повышению БА, другим фактором - сумма электронных плотностей в узлах сетки, приводящих к понижению БА. Тогда те узлы сетки, увеличение электронной плотности в которых приводит к повышению БА, можно отнести к фармакофорной части структуры, а узлы, увеличение электронной плотности в которых приводит к понижению БА, – к антифармакофорной части молекулы.
Классификация соединений на активные и неактивные с присвоением величин активностей Pk, равных соответственно 1 и 0, позволяет производить процедуру распознавания образов электронной структуры лекарственных средств с использованием алгоритма ConGO, что описано в п.п. 2.3. В этом случае количественная зависимость имеет вид функции желательности:
![]() , где
, где ![]()
Очевидно, что рассмотрение электронной структуры молекул требует оценки функции распределения электронной плотности в молекулах. Для этого могут быть использованы квантовохимические функции электронной плотности. Однако для сокращения временных затрат при расчетах в п.п. 2.4 предложен AlteQ подход. В данном алгоритме вклад электронной плотности атома А в молекулярную электронную плотность представляется в виде суммы (11) эмпирически подобранных экспоненциальных функций. Электронная плотность молекулы представляется в виде суммы (12) вкладов атомов в точке m молекулярного пространства:
![]() (11)
(11) 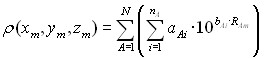 (12)
(12)
![]() и
и ![]() – параметры для каждого элемента Периодической системы, n – номер периода атома A, RA – расстояние от ядра атома А, N – общее количество атомов в молекуле. Выражение (11) предложено на основании анализа зависимостей логарифма экспериментальной электронной плотности (определенной прецизионным РСА) от расстояния до ядер атомов. Примеры подобных зависимостей представлены на Рис. 6 для атомов водорода и кислорода.
– параметры для каждого элемента Периодической системы, n – номер периода атома A, RA – расстояние от ядра атома А, N – общее количество атомов в молекуле. Выражение (11) предложено на основании анализа зависимостей логарифма экспериментальной электронной плотности (определенной прецизионным РСА) от расстояния до ядер атомов. Примеры подобных зависимостей представлены на Рис. 6 для атомов водорода и кислорода.
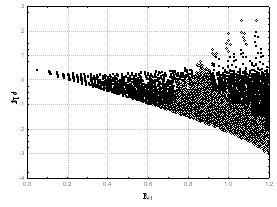
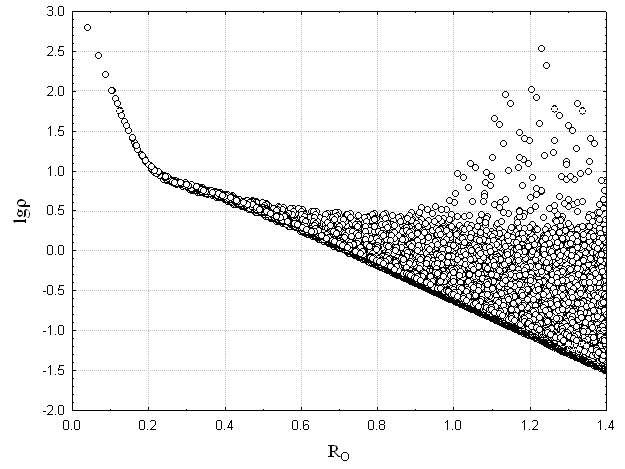
Рис. 6. Зависимость логарифма электронной плотности, определенной прецизионным РСА, от расстояния до центра ядра атомов Н, O в молекуле этокси-(2-меркапто-4,6-диметилгексагидропиримидин-5-ил)-метанола (соединение 1)
Нижняя часть данных графиков представляет собой логарифм электронной плотности атома без учета перекрывания с соседними атомами, более высокие значения отвечают областям перекрывания с соседними атомами. На данных графиках можно выделить участки, количество которых равно номеру периода, соответствующего рассматриваемому элементу. Поэтому количество функций в выражении (11) выбрано равным номеру периода, в котором находится рассматриваемый атом. Интегрирование AlteQ функции (11) электронной плотности по пространству равно числу электронов (NeA) в атоме A:
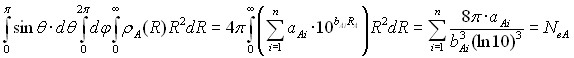
где ![]() и
и ![]() - параметры атома A, они были оценены на основании предположения о том, что каждая экспоненциальная функция i в выражении (11) должна описывать количество электронов (mAi) на квантовом уровне i. Тогда интегрирование по пространству функции i должно давать число электронов на соответствующем квантовом уровне.
- параметры атома A, они были оценены на основании предположения о том, что каждая экспоненциальная функция i в выражении (11) должна описывать количество электронов (mAi) на квантовом уровне i. Тогда интегрирование по пространству функции i должно давать число электронов на соответствующем квантовом уровне.
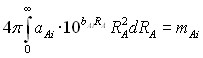 , при этом
, при этом ![]()
Параметры ![]() и
и ![]() представлены в Таблице 1 на примере элементов H, C, N, O, S, Cl. Обнаружено, что параметры атомов, соответствующие наиболее высокому квантовому уровню (i=n)
представлены в Таблице 1 на примере элементов H, C, N, O, S, Cl. Обнаружено, что параметры атомов, соответствующие наиболее высокому квантовому уровню (i=n) ![]() , отнесенные к радиусу максимальной электронной плотности (Rmax) коррелируют с потенциалом ионизации атома А (IA) с коэффициентом корреляции R=0.93 (S=2,0 эВ) (Рис. 7).
, отнесенные к радиусу максимальной электронной плотности (Rmax) коррелируют с потенциалом ионизации атома А (IA) с коэффициентом корреляции R=0.93 (S=2,0 эВ) (Рис. 7).
Это позволяет предположить то, что ![]() представляет собой эффективный заряд ядра, взятый с противоположным знаком. Сопоставление логарифма экспериментальной электронной плотности с логарифмом AlteQ-расчетной (
представляет собой эффективный заряд ядра, взятый с противоположным знаком. Сопоставление логарифма экспериментальной электронной плотности с логарифмом AlteQ-расчетной (![]() ) представлено на Рис. 8а,б на примере соединений 1 и ZrCl2 – (соединение 2). Для сравнения на Рис. 8в,г приведено аналогичное сопоставление, полученное при расчете электронной плотности квантовохимическими методами.
) представлено на Рис. 8а,б на примере соединений 1 и ZrCl2 – (соединение 2). Для сравнения на Рис. 8в,г приведено аналогичное сопоставление, полученное при расчете электронной плотности квантовохимическими методами.
Таблица 1
Параметры экспоненциальных AlteQ-функций
| NA | A | ||||||
| 1 | H | 3,1623 | -1,8672 | — | — | — | — |
| 6 | C | 848,42 | -9,5585 | 10,000 | -1,7265 | — | — |
| 7 | N | 1362,7 | -11,194 | 17,783 | -1,9418 | — | — |
| 8 | O | 2083,8 | -12,897 | 31,623 | -2,2138 | — | — |
| 9 | F | 2938,0 | -14,461 | 56,234 | -2,5478 | — | — |
| 16 | S | 16706 | -25,812 | 1000,0 | -6,3606 | 10,000 | -1,5083 |
| 17 | Cl | 18926 | -26,908 | 1584,9 | -7,4160 | 14,678 | -1,6283 |
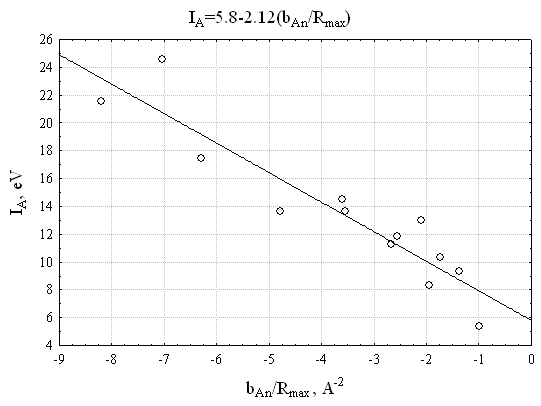 Рис. 7. Взаимосвязь потенциала ионизации IA и отношения Рис. 7. Взаимосвязь потенциала ионизации IA и отношения | Обнаружено, что величина |
около атомов водорода на расстояниях, превышающих ван-дер-ваальсовы радиусы атомов.
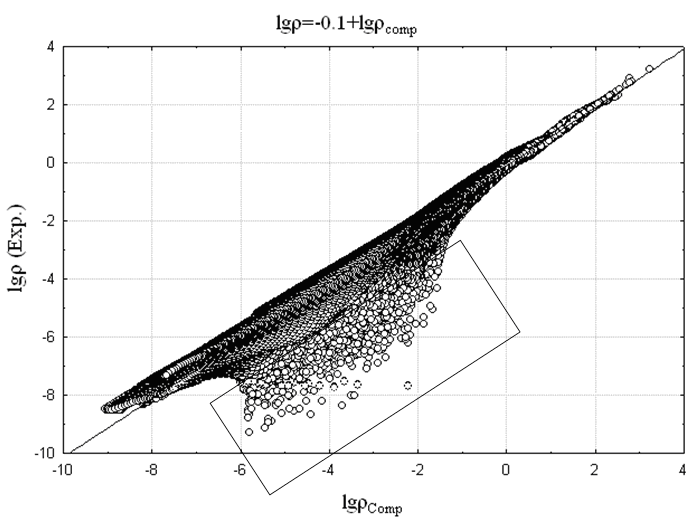
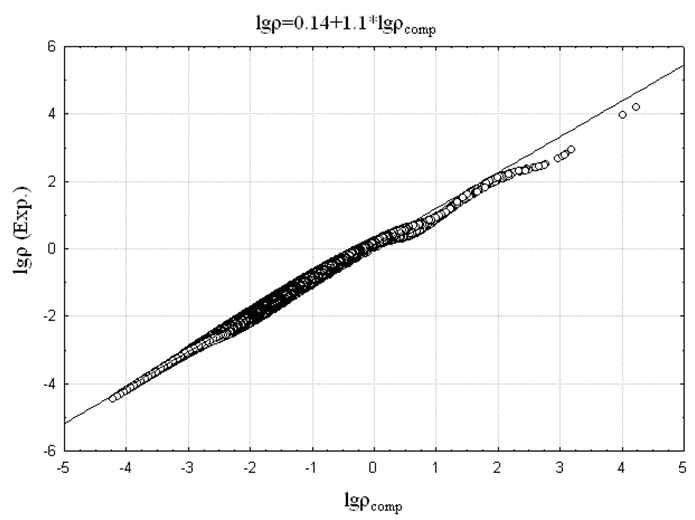
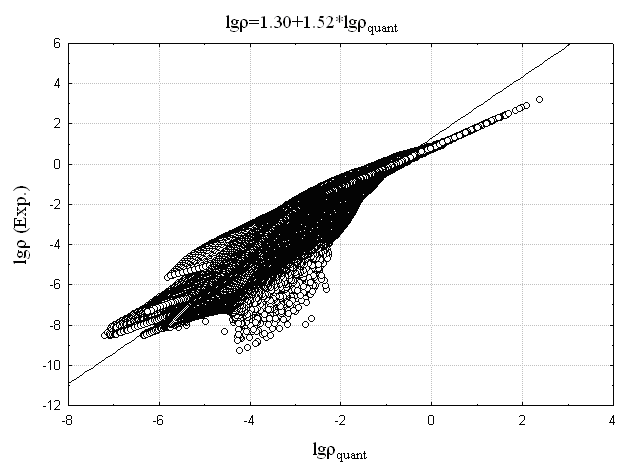
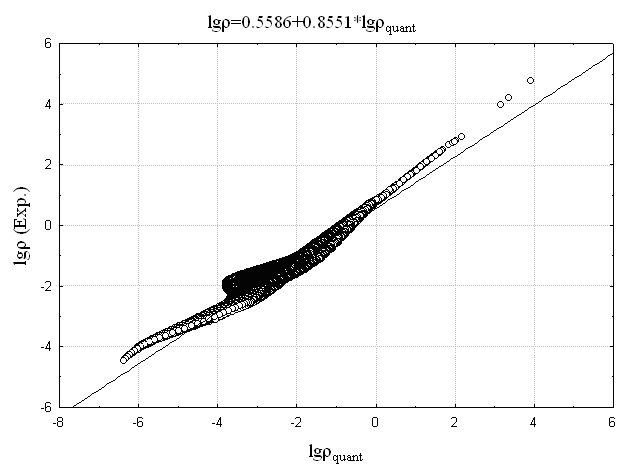
Рис. 8. Зависимость логарифмов экспериментальной и рассчитанной электронных плотностей в узлах сетки, найденной для: а) мол. 1 с помощью алгоритма AlteQ; б) мол. 2 с помощью алгоритма AlteQ; в) мол. 1 с помощью DFT B3LYP/6-311G(d,p); г) молекулы 2 с помощью HF, выбран базис 6-311G(d,p) для аппроксимации волновых функций атомов Cl и 3-21G – атома Zr.
Существование подобных точек связано с тем, что экспериментальное значение электронной плотности занижено благодаря ошибке вычитания вклада соседних молекул в кристалле. Кроме того, в соответствии с мультипольной моделью, интерпретация рефлексов на экспериментальной дифрактограмме осуществляется с использованием базисных квантовых функций слэйтерова типа. На Рис. 9б для сравнения показаны области наиболее сильного отклонения расчетных квантовохимических значений от экспериментальных значений электронной плотности для молекулы 1.
Таким образом, показано, что AlteQ может быть использован для корректного описания электронного строения молекул. Совокупное использование алгоритмов BiS/MC и ConGO позволяет полностью описать строение молекулы в момент взаимодействия с рецептором. Важнейшей стадией биологического действия лекарственных средств является их взаимодействие с целевым рецептором. Поэтому наиболее высоким уровнем рассмотрения биологически активных соединений является докинг соединений к рецептору, структура которого установлена с помощью РСА или ЯМР-спектроскопии.
![]()
![]()
Рис. 9. Области пространства в молекуле 1 с высокой ошибкой расчета электронной плотности: а) расчет выполнен алгоритмом AlteQ; б) квантовохимическим методом.
В п.п. 2.5 предложен метод ограниченного докинга, учитывающий основные недостатки существующих методов докинга. Данный подход основан на ориентации молекул в полости рецептора с использованием результата наложения биологически активных структур, полученного в рамках алгоритма BiS/MC с учетом всего комплекса взаимодействий в системе «рецептор-лиганд». Данные РСА комплекса “рецептор - лиганд“ хотя бы для одной молекулы выборки и установленная с помощью моделирования (алгоритм BiS/MC) их относительная ориентация, позволяют расположить молекулы всей выборки в реальном рецепторе (Рис. 10).
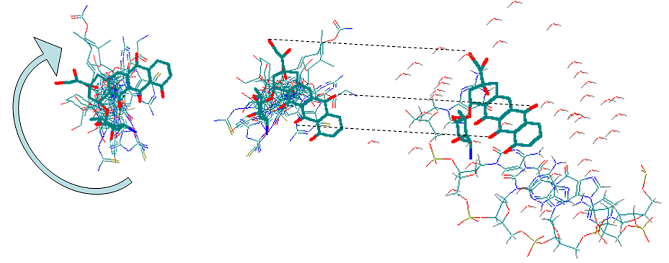
Рис. 10. Процедура ориентации молекул выборки в модельном рецепторе: а) поворот и
б) трансляция результата наложения структур, полученного с помощью алгоритма BiS/MC (жирным выделена молекула, входящая в состав экспериментально установленного комплекса),
в) комплекс, установленный РСА и используемый в качестве примера для ориентации
Для проверки качества ограниченного докинга молекул с использованием предложенного подхода в Protein Data Bank проведен отбор данных РСА более семидесяти комплексов рецепторов с лекарственными средствами. Среди них ДНК-антиметаболиты, ингибиторы p38 MAP-киназы, риновируса HRV14, термолизина, эластазы, циклин-зависимой киназы (CDK2) и дигидрофолатредуктазы. Результаты сравнения экспериментальной и расчетной ориентации показали, что точность докинга существенно превосходит имеющиеся аналоги. Так, в работе [Cosgrove D.A., Bayada D.M., Johnson A.P. A novel method of aligning molecules by local surface shape similarity// J. Comput.-Aided Mol. Design.- 2000.- 14.- P. 573 - 591] максимальное отклонение координат атомов (rMAX) истинной и расчетной ориентации по выборке ингибиторов риновируса HRV14 достигает 4.77 (соединение 1ro9), для ингибиторов термолизина - 11.14 (соединение 5tln), эластазы - 10.01 (соединение 1inc), для ингибиторов CDK2 - 18.70 (соединение 1ian), среднее отклонение координат атомов (RMS) истинной и расчетной ориентаций может превышать 9 (например, для соединения 1ian (ингибитор CDK2) RMS=9,72 ). При использовании алгоритма BiS для ориентации соединений тех же выборок RMS значительно менее 1, а rMAX по выборке, как правило, не превышают 2.
Для анализа полученных при докинге комплексов «рецептор-лиганд» в п.п.2.6 предложен новый алгоритм ConCon. Данный алгоритм позволяет определить зависимости БА соединений от параметров строения комплексов «рецептор-лиганд». Он основан на предположении о том, что БА вещества зависит главным образом от взаимодействия активных центров рецептора и лекарственного средства, все остальные атомы взаимодействующих частиц играют значительно менее важную роль в связывании благодаря конформационной или таутомерной вариабельности.
Для оценки энергии взаимодействия в алгоритме CoCon используется атом-атомный потенциал, включающий ван-дер-ваальсову и кулоновскую составляющие, оцениваемые с помощью модели MERA. Такое представление позволяет провести декомпозицию энергий парных взаимодействий в поиске наилучшей зависимости величины БА от параметров строения комплексов «рецептор-лиганд». Атомы рецептора и лиганда, чьи параметры входят в данную зависимость, предполагаются в алгоритме CoCon активными центрами. Декомпозиция реализуется в алгоритме CoCon путем использования пошагового включения и исключения факторов методами регрессионного описания с оценкой качества (CrossR2) моделей по технике «скользящий контроль». Поскольку количество параметров для построения регрессионной модели должно быть одинаковым, в алгоритме CoCon предложены следующие подходы. Первый подход: рассматривается взаимодействие всей молекулы лекарственного средства с активными центрами рецептора. Данный подход реализуется в CoCon построением зависимостей (13-15):
![]() (13)
(13) ![]() (14)
(14) ![]() (15)
(15)
где a, b, c – параметры; ![]() - энергии взаимодействия i-того активного центра рецептора со всеми атомами лиганда;
- энергии взаимодействия i-того активного центра рецептора со всеми атомами лиганда; ![]() силы взаимодействия j-того активного центра рецептора со всеми атомами лиганда,
силы взаимодействия j-того активного центра рецептора со всеми атомами лиганда, ![]() - энергия взаимодействия i-того активного центра рецептора со всеми атомами лиганда,
- энергия взаимодействия i-того активного центра рецептора со всеми атомами лиганда, ![]() - энергия взаимодействия j-того активного центра молекул воды со всеми атомами лиганда.
- энергия взаимодействия j-того активного центра молекул воды со всеми атомами лиганда.
Рассмотрение сил в выражении (13) позволяет осуществить учет динамики взаимодействия в системе «рецептор-лиганд», поскольку сила является векторной величиной, показывающей направление движения молекулы в результате взаимодействия. Силы в алгоритме CoCon определяются с использованием выражения (10).
Второй подход: молекула каждого лиганда разбивается на фиксированное количество фрагментов, при этом рассматриваются взаимодействия атомов рецептора не со всей молекулой, а с каждым фрагментом. Количество фрагментов одинаково для всех молекул выборки, в результате чего число факторов для построения регрессионных моделей также одинаково. Разбиение молекулы на фрагменты осуществляется с постоянным телесным углом, как показано на Рис.11. Далее строятся уравнения (16) и (17) с использованием регрессионного анализа:
![]() (16)
(16) ![]() (17)
(17)
где ![]() - энергия взаимодействия всех атомов рецептора (N(R) – общее количество атомов сайта рецептора) с l-тым фрагментом лиганда,
- энергия взаимодействия всех атомов рецептора (N(R) – общее количество атомов сайта рецептора) с l-тым фрагментом лиганда, ![]() - энергия взаимодействия всех молекул воды сайта рецептора (общее количество молекул воды N(W)) с k-тым фрагментом лиганда,
- энергия взаимодействия всех молекул воды сайта рецептора (общее количество молекул воды N(W)) с k-тым фрагментом лиганда, ![]() - энергия взаимодействия i-того активного центра белковой части рецептора
- энергия взаимодействия i-того активного центра белковой части рецептора
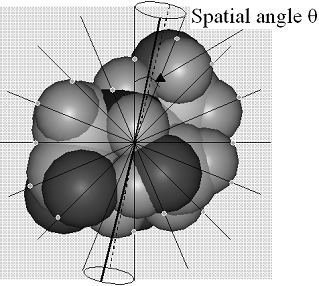 Рис. 11. Определение точек на поверхности молекулы для вычисления параметров взаимодействия с сайтом. Рис. 11. Определение точек на поверхности молекулы для вычисления параметров взаимодействия с сайтом. | с j-тым фрагментом лиганда, |
В подавляющем большинстве случаев водородная связь характеризуется расстоянием, равным половине суммы расстояний ковалентного и ван-дер-ваальсова контакта. Более того, она имеет энергию, большую по сравнению с энергией ван-дер-ваальсовых взаимодействий и меньшую по сравнению с энергией ковалентных взаимодействий. На основании этих данных вычисляются параметры a, b, c и выражений (18)-(20), что может быть представлено в виде систем уравнений (18а), (19а), (20а), описанных ниже. Подобный расчет параметров делает оценку энергий водородных связей неэмпирической.
Первый вариант расчета энергий водородных связей (EH) представлен выражением, аналогичным функции распределения Кирквуда (Рис. 12):
![]() (18)
(18)
где a, b, c и – его параметры, определяемые из следующей системы уравнений:
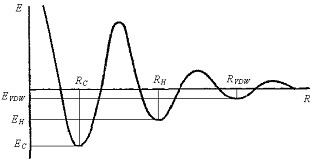 Рис. 12. Функция для оценки энергий водородных связей, аналогичная функции распределения Кирквуда. Рис. 12. Функция для оценки энергий водородных связей, аналогичная функции распределения Кирквуда. | 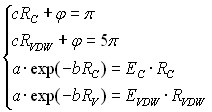 (18а) где RC – равновесное расстояние ковалентного связывания, EC – энергия ковалентного связывания, RVDW - равновесное расстояние ван-дер-ваальсового контакта, EVDW – его энергия, RH – равновесное расстояние водородной связи. (18а) где RC – равновесное расстояние ковалентного связывания, EC – энергия ковалентного связывания, RVDW - равновесное расстояние ван-дер-ваальсового контакта, EVDW – его энергия, RH – равновесное расстояние водородной связи. |
Другой вариант расчета энергий водородных связей представлен:
![]() (19)
(19)
Параметры a и b определяются в CoCon, исходя из предположения о том, что EH соответствует минимуму энергии взаимодействия. Поэтому параметры могут быть найдены из следующей системы уравнений:
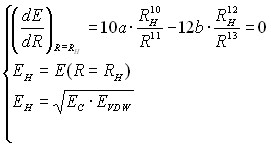 или
или 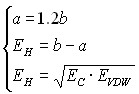 (19а)
(19а)
Третий вариант представлен как:
![]() (20)
(20)
Параметры функции рассчитываются аналогично предыдущему предположению из следующей системы уравнений:
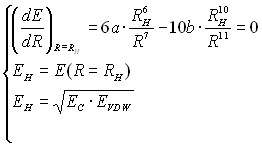 или
или 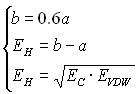 (20а)
(20а)
Таким образом, CoCon формирует 5 вариантов зависимостей биологической активности от параметров строения комплексов «рецептор-лиганд» (выражения (13)-(17)). Их анализ позволяет выделить наиболее важные для взаимодействия в комплексах «рецептор-лиганд» фрагменты в структуре белка, молекул воды сайта рецептора, лиганда. CoCon определяет наиболее важные типы межмолекулярных взаимодействий: электростатических, ван-дер-ваальсовых и водородных связей. Для расчета энергий водородных связей в CoCon используется 3 новых подхода. Использование совокупности алгоритмов BiS/MC, ConGO и CoCon позволяет наиболее полно описать молекулу биологически активного соединения и рецептора в момент взаимодействия, определить количественные взаимосвязи БА с параметрами строения молекул и их комплексов с рецептором. Каждый из этих подходов может быть использован как индивидуально, так и в комбинации друг с другом, что повышает прогностическую ценность полученных количественных моделей.
В главе 3 рассматриваются примеры теоретического исследования различных выборок биологически активных соединений. В п.п. 3.1 рассмотрено 24 противоопухолевых ингибитора дигидрофолатредуктазы (DHFR) из базы данных Национального института рака. Некоторые из них представлены на схеме 1.
Схема 1.
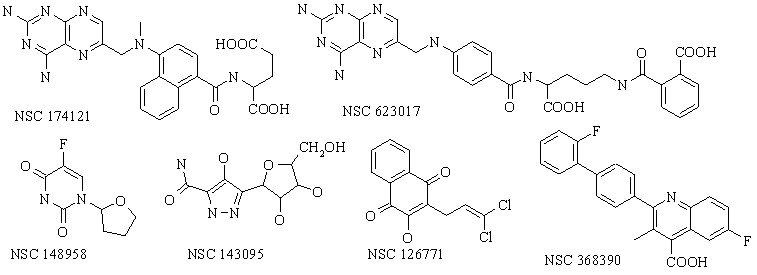
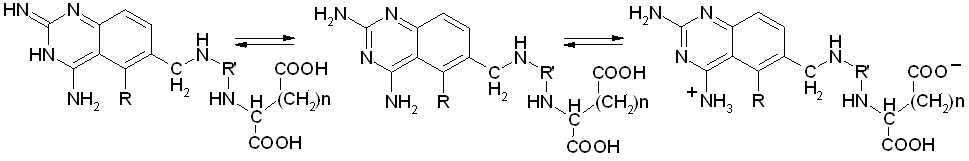
Данные соединения могут существовать в различных ТК формах. Примеры таутомерных переходов представлены на Схеме 2. Всего для данной выборки соединений было рассмотрено около 22000 ТК форм.
Схема 2.
Анализ в рамках алгоритма BiS/MC позволил определить самые активные ТК формы для каждого ингибитора. Показано, что ими, как правило, являются цвиттер-ионные формы соединений, имеющие развернутую структуру, обеспечивающую эффективное взаимодействие со всеми атомами модельного рецептора (Рис. 13). При этом термодинамически более выгодной ТК
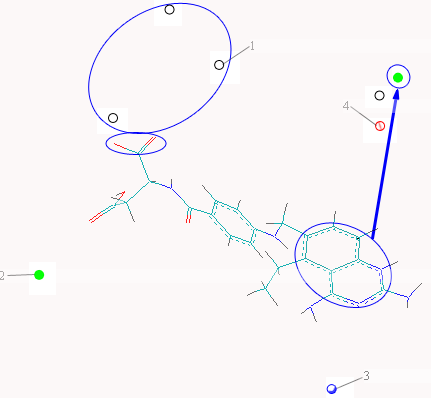 Рис.13. Ориентация наиболее биологически активных ингибиторов в полости модельного рецептора на примере ингибиторов: А) NSC 184692. 1 – H, 2 – C, 3 N, 4 – O. Рис.13. Ориентация наиболее биологически активных ингибиторов в полости модельного рецептора на примере ингибиторов: А) NSC 184692. 1 – H, 2 – C, 3 N, 4 – O. | формой являются свернутые структуры. Сопоставление расчетных активных ТК форм с найденными экспериментально представлено на Рис. 3а на примере комплекса 1hfp. Данный пример демонстрирует хорошее согласие расчетной ТК формы экспериментальным данным. Обнаружено, что ТК формы, определяющие связывание наиболее биологически активных ингибиторов, имеют большое количество открытых атомов кислорода за счет развернутой структуры, что позволяет им эффективно взаимодействовать с атомами водорода модельного рецептора (Рис.13). |
В рамках 3D QSAR алгоритма ConGO проведен детальный анализ электронной структуры наиболее активных ТК форм, определяющих действие соединений в рецепторе. Показано, что в случае наиболее активных молекул антифармакофорные фрагменты электронной структуры находятся на расстояниях, как правило, превышающих ван-дер-ваальсовы радиусы атомов (Рис. 14а). Электронная плотность на данных расстояниях чрезвычайно низка, поэтому ее негативное влияние на величину активности для высокоактивных соединений сведено к минимуму.
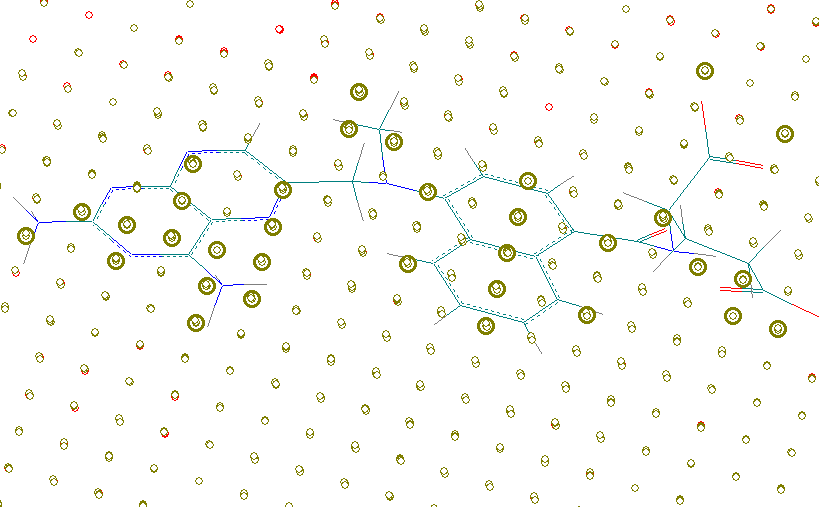
Рис. 14. Фармакофорные (желтые точки) и антифармакофорные (красные точки) фрагменты ингибиторов DHFR: а) NSC 174121 (pGI50%=8,20), б) NSC 19893 (pGI50%=4,59). Жирным выделены точки, расположенные в пределах ван-дер-ваальсовых радиусов атомов
Точки, определяющие фармакофорные фрагменты, расположены в основном в области ароматических колец, атомов азота и открытых атомов кислорода. Фармакофорное влияние ароматических колец у наиболее активных молекул связано с их эффективными -стэкинговыми взаимодействиями с ароматическими системами фермента. Атомы азота и атомы кислорода обеспечивают образование водородных связей и электростатических взаимодействий с полярными фрагментами фермента. Для неактивных соединений наблюдаются в основном точки с антифармакофорным влиянием (Рис. 14б).
Использование данных РСА комплекса 1rg7 позволило осуществить докинг молекул выборки к полости DHFR. Показано, что высокоактивные молекулы проникают глубоко в полость DHFR, тогда как малоактивные молекулы остаются у входа в полость, что препятствует их эффективному взаимодействию с сайтом. Аминокислотные остатки Ala19, Asn23, Leu28, Asp27, Phe31, Thr46, Ile50, Arg52 и Ile94 образуют значительное количество сокращенных контактов и водородных связей с ингибиторами. Показано, что величина активности pGI50% связана с энергией водородных связей (выражение (19)) с R=0.756 (F=21, S=0.88).
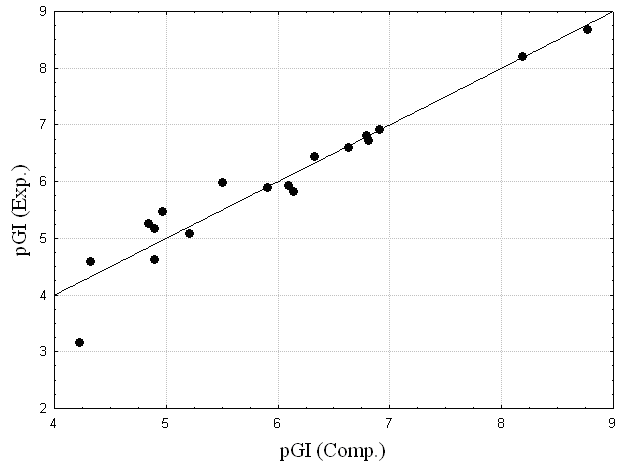 Рис. 15. Экспериментальные и расчетные pGI50% ингибиторов DHFR Рис. 15. Экспериментальные и расчетные pGI50% ингибиторов DHFR | Наилучшая зависимость, построенная с использованием алгоритма CoCon, описывает величину активности ингибиторов DHFR с CrossR2=0.91 (R=0.96, S=0.10) (Рис.15): 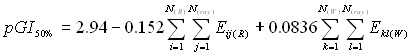 Критическими аминокислотными остатками являются Ile5, Asn23, Pro25, Leu28, Trp30, Phe31, Lys32, Gly43, Arg44, Ser49, Ile50, Gly51, Leu 54, Ile94 и Gly96, обеспечивающие короткие контакты с лигандами. Критическими аминокислотными остатками являются Ile5, Asn23, Pro25, Leu28, Trp30, Phe31, Lys32, Gly43, Arg44, Ser49, Ile50, Gly51, Leu 54, Ile94 и Gly96, обеспечивающие короткие контакты с лигандами. |
В п.п. 3.2 исследованы ингибиторы нейраминидазы, подавляющее большинство которых представляет собой органические соли натрия, калия, аммониевых оснований, пиридиния. Поскольку в выборку соединений входят соли, поэтому для моделирования их оптимальной
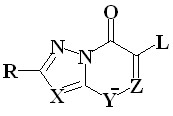 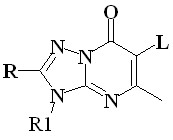 Кat+ Кat+ = Na+, K+, аммониевые катионы, пиридиний X, Z = N, C; Y = N-; R = Me, H, Sme, Ph, Py; R1=Me, C12H25, C4H8Oаc Кat+ Кat+ = Na+, K+, аммониевые катионы, пиридиний X, Z = N, C; Y = N-; R = Me, H, Sme, Ph, Py; R1=Me, C12H25, C4H8Oаc | геометрии с учетом влияния водного окружения, составляющего основу внутренней среды организма человека, использован алгоритм MOPS, осуществляющий поиск наиболее выгодных комплексов «катион-анион» вдоль колебательных мод гессиана с континуальным учетом влияния растворителя. Примеры структур представлены на Рис. 16. |
В рамках 3D-QSAR алгоритма BiS/MC определены структуры, определяющие связывание ингибиторов с нейраминидазой, проведено построение комплексов псевдоатомного рецептора с ингибиторами, найдено уравнение зависимости противовирусной активности от параметров строения комплексов «модельный рецептор - лиганд» с CrossR2=0.999.
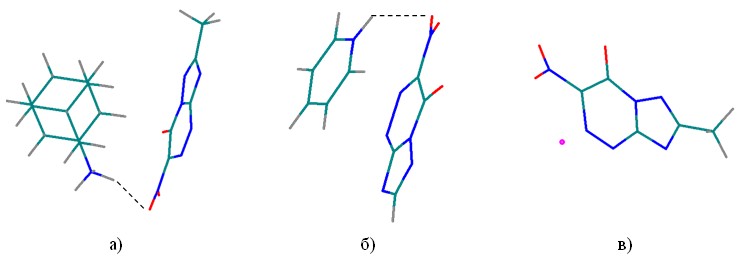
Рис. 16. Геометрия наиболее термодинамически выгодных структур солей – ингибиторов нейраминидазы
Рассмотрение вида комплексов «модельный рецептор – лиганд» выявило следующие закономерности. Обнаружено, что катионы наиболее активных соединений ориентируются рядом с отрицательно заряженной частью модельного рецептора, а анионы - рядом с положительно заряженной частью рецептора (Рис. 17а). Для менее активных соединений характерна обратная картина: катион образует невыгодные электростатические взаимодействия с положительно заряженной частью модельного рецептора, представленной атомами водорода, а анион – с электроотрицательными атомами азота и кислорода (Рис.17б).
Проведено распознавание образов электронной структуры ингибиторов нейраминидазы в рамках алгоритма ConGO. Примеры соединений с фармакофорными и антифармакофорными узлами сетки ConGO представлены на Рис.18. Обнаружено, что активные соединения характеризуются большим количеством фармакофорных фрагментов по сравнению с малоактивными структурами, многие из фармакофорных фрагментов приходятся на катионную часть ингибиторов (Рис. 18), локализуясь в пределах ван-дер- ваальсовых радиусов атомов.
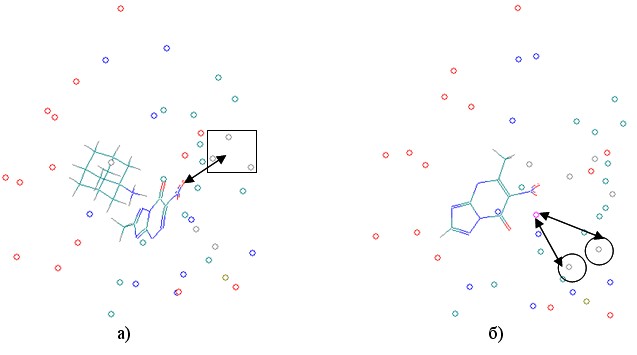
Рис. 17. Комплексы ингибиторов нейраминидазы с псевдоатомным рецептором (BiS/MC)
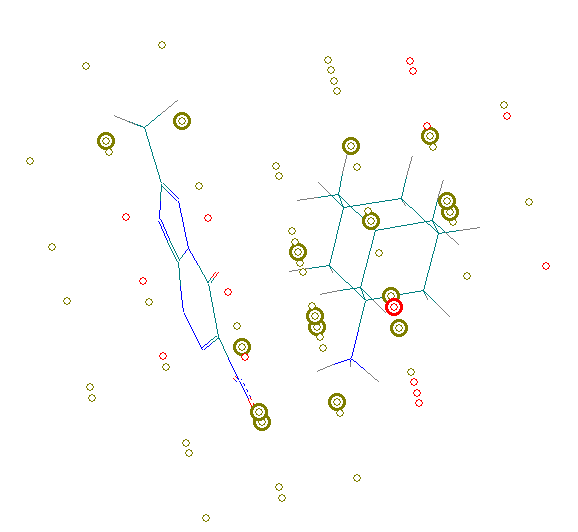 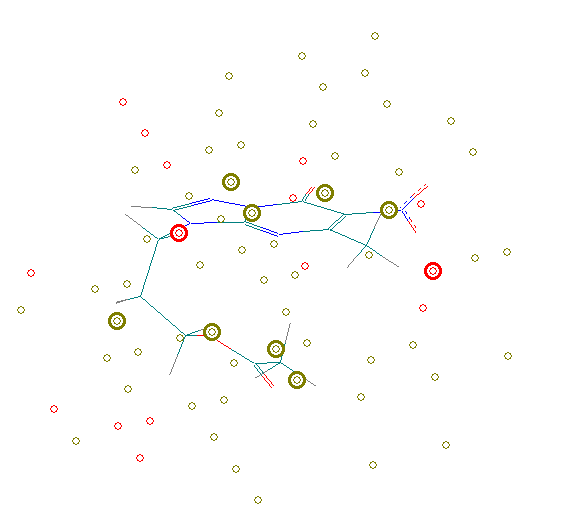 Рис. 18. Фармакофорные и антифармакофорные фрагменты электронной структуры ингибиторов нейраминидазы: а) активное соединение (Ef=91%); б) малоактивное (Ef<1%). Рис. 18. Фармакофорные и антифармакофорные фрагменты электронной структуры ингибиторов нейраминидазы: а) активное соединение (Ef=91%); б) малоактивное (Ef<1%). | Показано, что анализ электронной структуры соединений позволяет распознавать ингибиторы нейраминидазы с качеством, определенным по технике «скользящий контроль», равным 100%. Проведен докинг соединений к полости нейраминидазы с использованием комплекса 2gwh, для которого в Protein Data Bank найдены данные РСА. |
Рассмотрение структур полученных комплексов «рецептор-лиганд» показало, что полость, в которую встраивается ингибиторы, представлена Arg118, Glu119, Ile149, Asp151, Arg152, Glu196, Asn221, Ile222, Arg224, Glu227, Ala246, Glu276, Glu277, Lys292, Arg371, Tyr406, Pro431. Поверхность полости содержит большое количество NH2, COOH, CO, OH-групп. Высокая гидрофильность полости приводит к координации значительного числа молекул воды, которые принимают активное участие в стабилизации комплекса «фермент - лиганд». Для наиболее активных соединений наблюдается более эффективное связывание с данными аминокислотными остатками: отрицательно заряженные атомы/группы атомов ингибитора координируются около положительно заряженных атомов водорода; положительно заряженная часть соединений стремиться расположиться рядом с электроотрицательными атомами –NН-, -СОО-, -ОН групп фермента, что хорошо согласуется с результатами моделирования псевдоатомного рецептора в рамках алгоритма BiS/MC. При этом образуется максимальное количество электростатических взаимодействий. У менее активных соединений наблюдаются обратные закономерности в строении комплексов. Кроме того, было показано, что важным для проявления высокой активности является способность катиона встраиваться в полость рецептора, взаимодействовать атомами водорода NH2-группы с отрицательно заряженными атомами кислорода Asp151 фермента, образовывать дополнительное ван-дер-ваальсовое взаимодействие метиленовых фрагментов адамантиламмония, либо атомов водорода и углерода кольца пиридиния с атомами водорода метильных и метиленовых групп аминокислот Ile222, Arg224, Ala246 фермента. Высокая способность катиона к гидратации не позволяет ему проникать в полость фермента, что понижает активность солей натрия и калия. В рамках алгоритма CoCon получена зависимость эффективности (Ef) ингибиторов от энергии взаимодействий с белковой частью рецептора и водным окружением (CrossR2=0,92, R=0,97, S=2,6.):
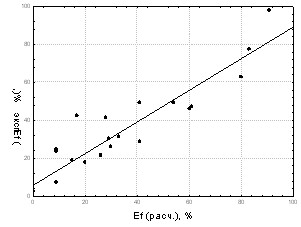 Рис. 19. Экспериментальная и расчетная (определенная с использованием CoCon) активности ингибиторов нейраминидазы Рис. 19. Экспериментальная и расчетная (определенная с использованием CoCon) активности ингибиторов нейраминидазы | 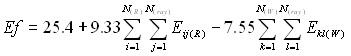 График зависимости представлен на Рис.19. Одной из важных проблем при создании лекарственных средств является создание препаратов, селективно действующих на нужную биомишень. Так, например, в работе [S. Guccione, A.M. Doweyko, H. Chen, G.U. Barretta, F. Balzano., J. of Comp.-Aid. Mol. Des., 2000.-V.14.-p. 647] изучены активности соединений в отношении конкурирующих 5-HT1A и 1-AR рецепторов. График зависимости представлен на Рис.19. Одной из важных проблем при создании лекарственных средств является создание препаратов, селективно действующих на нужную биомишень. Так, например, в работе [S. Guccione, A.M. Doweyko, H. Chen, G.U. Barretta, F. Balzano., J. of Comp.-Aid. Mol. Des., 2000.-V.14.-p. 647] изучены активности соединений в отношении конкурирующих 5-HT1A и 1-AR рецепторов. |
Поэтому в п.п. 3.3 проведен детальный анализ электронной структуры данных соединений в рамках алгоритма ConGO с распознаванием образов селективно действующих ингибиторов 5-HT1A и 1-AR рецепторов. Показано, что активные соединения в обоих случаях распознаются
 | с качеством, определенным по технике «скользящий контроль», составляющим 100%. Рассмотрение действующих ТК форм показало, что примерно в 50% случаев связывание с данными рецепторами определяют различные зеркально противоположные конформеры. В остальных 50% случаев действующим на этих двух рецепторах оказывается |
один и тот же конформер. При этом для обоих рецепторов в случае активных соединений было обнаружено преимущественное действие конформеров, характеризующихся компактной геометрией (см. Рис. 20а), позволяющей им без труда встраиваться в полости. У неактивных соединений связывание с данными рецепторами определяют конформеры, имеющие развернутое строение (Рис. 20б), приводящее к большим размерам молекулы, что может препятствовать встраиванию в полости 5-HT1A и 1-AR рецепторов.
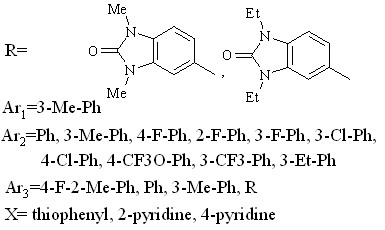
Рассмотрение отличий в проявлении ингибирующего действия на 5-HT1A и 1-AR рецепторы показало, что серьезная разница наблюдается в антифармакофорных частях молекул при их действии на данные рецепторы. Так, на Рис. 20б на примере одного из ингибиторов пунктиром выделены антифармакофорные фрагменты при действии данного соединения на 5-HT1A рецептор, сплошной линией – на 1-AR рецептор. Наибольшая разница в величинах pIC50% при ингибировании 5-HT1A и 1-AR рецепторов наблюдается для соединения, представленного на Рис.20в. Поэтому оно было рассмотрено в качестве эталонного для рекомендаций к усилению селективного действия по отношению к 5-HT1A рецептору.
Рис. 20. Связывающиеся с 5-HT1A и/или 1-AR рецепторами конформеры: а) активного соединения, б) малоактивного соединения, в) соединения, наиболее селективно действующего на 5-HT1A-рецептор
Обнаружено, что данное соединение не имеет антифармакофорных фрагментов при действии на 5-HT1A рецептор. При взаимодействии с 1-AR рецептором наблюдаются антифармакофорные фрагменты, показанные на Рис. 20в пунктиром. Поэтому для понижения активности данного соединения по отношению к 1-AR рецептору в данные области следует ввести более тяжелые атомы. Так, например, заменить помеченные крестом атомы водорода на атомы галогенов. Данные замены приведут к усилению электронной плотности в антифармакофорной части соединения по отношению 1-AR рецептору, а значит понижению активности в отношении данного рецептора. Кроме того, подобные замены должны усилить ингибирующее действие на 5-HT1A рецептор, поскольку они лишь усилят фармакофорную часть молекулы по отношению к данному рецептору.
В п.п 3.4. проведен анализ БА ингибиторов p38 MAP киназы в мультитаутомерно-конформационном режиме, рассмотрено более 280 ТК форм для 24 ингибиторов, для которых в литературе найдены величины IC50% (моль/л). Для данных соединений было обнаружено серьезное отличие в величинах активностей для разных энантиоконформеров. Так, например, энантиоконформер, представленный на Рис. 21а, является наиболее активным, в большей степени определяющим БА всего соединения, расчетная величина активности для него составляет pIC50% (comp.)=31,92, вероятность существования составляет 0,111.
 |  |
Его равновероятный зеркальный антипод - энантиоконформер, представленный на Рис. 21б, является наименее активным, он характеризуется расчетной величиной активности, равной pIC50% (comp.)= -15,46. В целом pIC50% для данного соединения составляет 7,52. Взаимосвязь экспериментальной и расчетной БА соединений, найденная в рамках алгоритма BiS/MC, представлена на Рис. 21в.
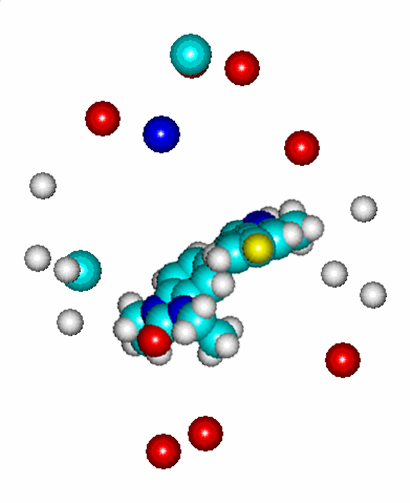
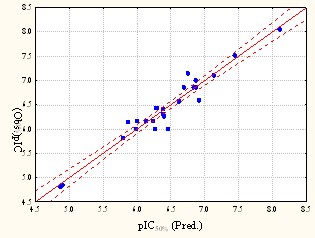
Рис. 21. Ингибиторы р38 МАР-киназы: а) и б) ориентация двух энантиомеров в модельном рецепторе (BiS/MC), в) взаимосвязь экспериментальной и расчетной БА (BiS/MC).
Величина активности ингибиторов р38 МАР киназы была определена in vitro, однако в живом организме данные соединения могут метаболизировать. Важную роль в метаболизме соединений эндогенного и экзогенного происхождения играют различные изоформы цитохрома Р450, механизмы метаболизма на которых рассмотрены, например, в п.п. 3.5-3.7. Полученные обучающие правила были использованы для прогноза возможности метаболизма ингибиторов р38 МАР киназы. Было обнаружено, что все изученные соединения будут метаболизировать с высокой вероятностью на 2D6 и 3A7 изоформах, и подавляющее большинство – на различных изоформах 3A подсемейства. Таким образом, данные соединения с большой вероятностью не достигнут р38 МАР киназы в клетке, метаболизируя в ней.
В п.п. 3.5-3.8 рассмотрены субстраты изоформ цитохрома Р450, для которых в литературе найдены константы Михаэлиса KM (моль/л), изучены детали механизмов метаболизма на разных изоформах с целью получения обучающих правил для прогноза метаболизма новых соединений. В п.п. 3.5 рассматриваются субстраты изоформы 3А4 (примеры показаны на Схеме 3), для которых в литературе найдены константы Михаэлиса (ммоль/л), отражающие эффективность метаболизма. Предварительный анализ с использованием нейронных сетей и дискриминантного анализа показал, что для субстратов 3А4 изоформы характерна молекулярная масса Mr=200448 г/моль и минимальный размер вдоль главной оси вращения, равный 4.68-8.00. Возможно, это обеспечивает встраивание соединений в полость 3А4 изоформы, приводя к возможности их метаболизма. Однако эти параметры не являются единственным условием проявления активности в отношении рассмотренной изоформы, поскольку качество распознавания субстратов 3А4 изоформы при таком рассмотрении не превышает 70%.
Схема 3.
ацетаминофен Индинавир ритонавир амброксол
таксотер ретиналь тирилазад
Поэтому проведен более детальный анализ механизмов реакций метаболизма соединений на 3А4 изоформе с рассмотрением взаимодействий в системе "рецептор-лиганд". Для каждого соединения выборки проведен мультиконформационный анализ. Рассмотрение найденных конформеров показало, что число наиболее вероятных конформеров у соединений выборки может колебаться от 1 до нескольких десятков. Так, например, для таких субстратов, как мидазолам, салициловая кислота, 2-хлоро-3-пиридин-3-ил-5,6,7,8-тетрагидроиндолизин-1-карбоксамид, включающих циклы, имеющих от 1 до 4 связей вращения количество вероятных конформеров составляет 2-4. Для таких соединений, как целекоксиб, иринотекан, зипрасидон, имеющих значительное число связей вращения, найдено соответственно 28, 12, 44 конформера. Наибольшее число конформеров наблюдается в случае делавирдина, их число достигает 68. Ряд соединений, например, пропафенон, бупивакаин, бупренорфин, несмотря на значительное число связей вращения (9-13), может существовать всего лишь в виде 1-4 конформеров. Связано это с образованием в подобных структурах эффективных внутримолекулярных взаимодействий, например водородных связей, которые препятствуют вращению. Полученные мульти-конформационные модели молекул рассмотрены с использованием 3D/4D QSAR
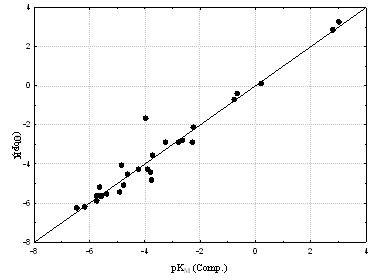 Рис. 22. Расчетные и экспериментальные величины показателей констант Михаэлиса (N=27, R=0,91, CrossR2=0,88, S=0,17) Рис. 22. Расчетные и экспериментальные величины показателей констант Михаэлиса (N=27, R=0,91, CrossR2=0,88, S=0,17) | алгоритма BiS/MC. Качество полученной количественной модели составило CrossR2=0,88. Экспериментальные и расчетные величины pKM показаны на Рис. 22. Рассмотрение расчетных активностей конформеров показало, что большинство субстратов метаболизирует в конформациях, энергия которых выше энергии структуры, соответствующей глобальному минимуму. Кроме того, было найдено, что для ряда соединений энантиоконформеры |
характеризуются значительным отличием в эффективности метаболизма. Так, для галоперидола, бупивакаина конформеры, характеризующиеся максимальным и минимальным показателем константы Михаэлиса, являются зеркальными антиподами. В случае рацемического оксибутинина метаболизму на 3А4 изоформе подвергается в большей степени S-стереоизомер (pKM (max)=10.48), R-стереоизомер характеризуется показателем константы Михаэлиса, не превышающим 1.53. Более активными являются компактные ТК формы.
Анализ полученных модельных комплексов с субстратами выявил, что для активных соединений наблюдается: 1) неэффективное связывание малополярных групп субстрата, например, ароматических колец, с областью I (Рис.23а), представленной заряженными атомами кислорода и водорода, и 2) эффективное связывание положительно заряженных атомов водорода с областью II, представленной атомами кислорода и азота. Для наименее активных структур наблюдаются обратные закономерности (Рис. 23б). Структура модельных комплексов позволяет предположить, что область I может имитировать молекулы воды. Тогда выгодное связывание субстрата с данными атомами может препятствовать встраиванию в изоформу 3А4, которую могут имитировать атомы области II.
Проведен докинг отобранных наиболее активных конформеров субстратов в полости изоформы с использованием данных РСА ее комплекса с эритромицином (2j0d). Рассмотрение расчетных комплексов позволило выделить значительное количество липофильных H…H сокращенных контактов между молекулой субстрата и атомами сайта изоформы. Такие контакты
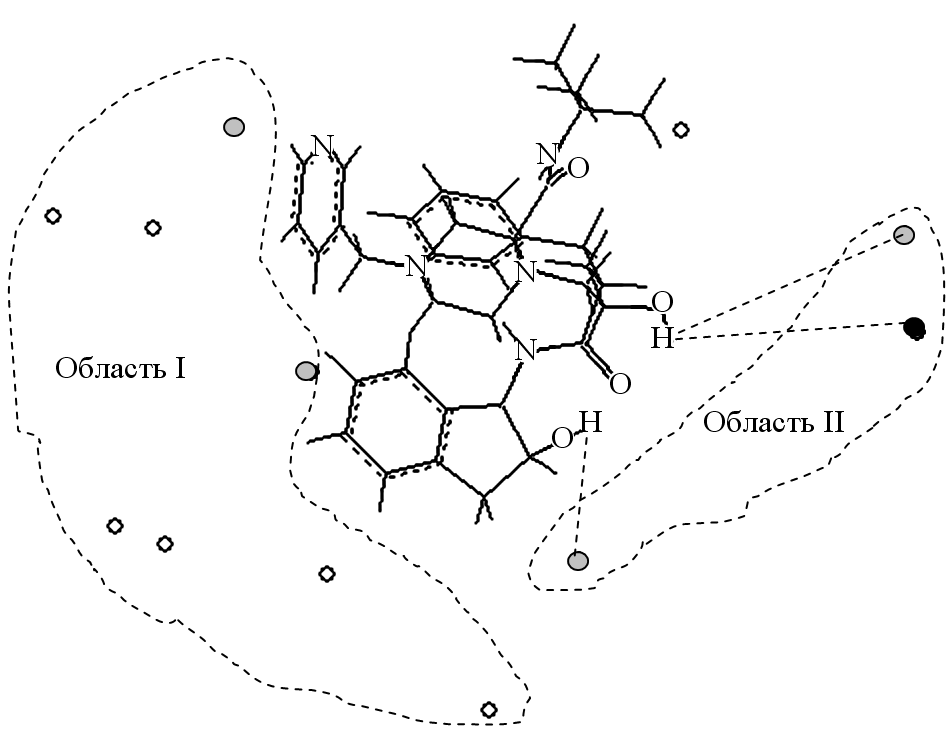
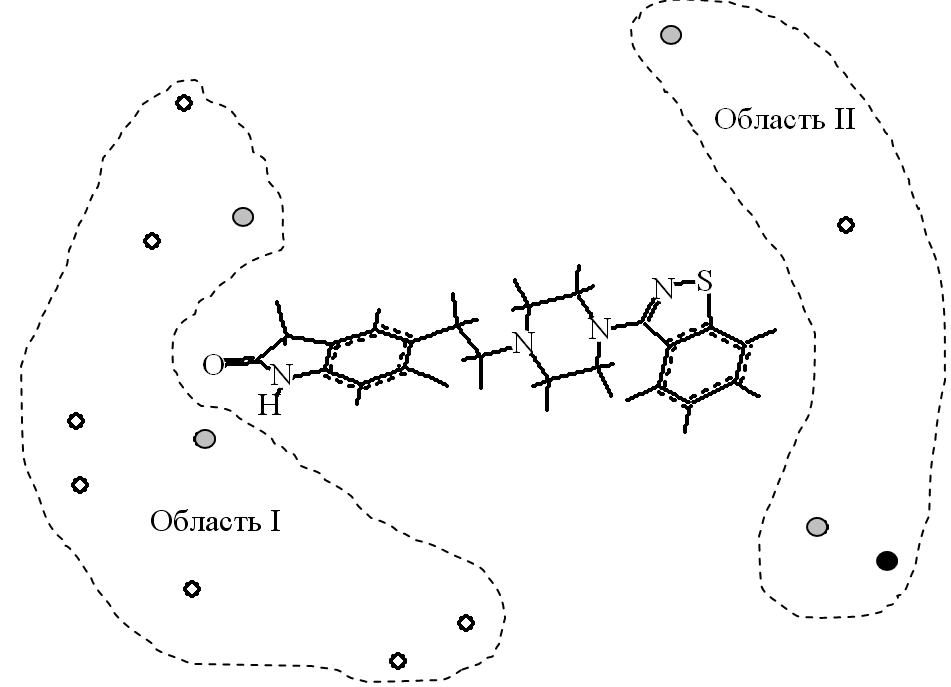
Рис.23. Модельные комплексы субстратов с изоформой: а) для наиболее активной структуры – конформера индинавира (pKM=11.55). б) наименее активной структуры – конформера зипрасидона (pKM=-33.61). - атомы водорода, - атомы кислорода, - атомы азота
являются преимущественными даже в случае молекул с существенным количеством потенциальных центров образования водородных связей. Принято считать, что метаболизм соединений осуществляется под действием молекулы кислорода, связанной с атомом железа гема. Однако рассмотрение комплексов показало, что в подавляющем большинстве случаев метаболизируемые реакционные центры субстратов локализованы от атома железа гема изоформы на расстояниях, превышающих 8. Кроме того, данные РСА показывают отсутствие молекулы кислорода в комплексе 2j0d. Поэтому предположено, что метаболизм субстратов осуществляется с участием атома железа гема по эстафетному механизму посредством молекул воды, заполняющих полость, либо с участием аминокислотных остатков полости (например, Arg105), образующих в ряде случаев водородные связи с метаболизируемыми центрами субстратов.
Обнаружено, что важным для повышения pKM является эффективное взаимодействие субстратов с атомом углерода Phe220. С использованием алгоритма CoCon найдено уравнение зависимости pKM от энергии взаимодействия с данным атомом (R=0.774, S=0.15).
![]()
Данный аминокислотный остаток локализован у входа в полость изоформы, поэтому молекулы больших размеров (индинавир, таксотер), заполняющие всю полость изоформы, способны образовывать межмолекулярные C…H липофильные взаимодействия с данным атомом. Расстояние между контактирующими атомами в таких случаях составляет 2.3-2.6.
Наилучшая количественная модель, найденная с использованием CoCon, описывает pKM с CrossR2=0,92 (R=0,97, S=0,05):
![]()
Атомы, параметры взаимодействия с которыми входят в данную зависимость, а также критические аминокислотные остатки, включающие данные атомы, показаны на Рис.24.
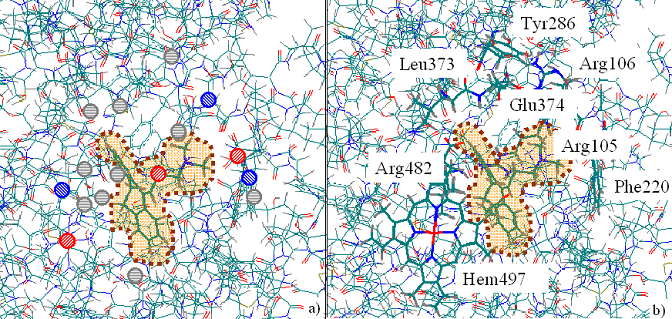
Рис. 24. Активные центры и критические аминокислотные остатки 3А4 изоформы.
Другой наиболее распространенной изоформой цитохрома P450 является 1A2 изоформа. Поэтому в п.п.3.6. исследован механизм метаболизма 17 субстратов данной изоформы, для которых в литературе найдены направления реакций метаболизма. Примеры данных соединений представлены на схеме 4. Для каждого из них проведен ТК анализ, построен псевдоатомный рецептор с использованием алгоритма BiS/MC, выделены наиболее активные ТК формы, определяющие метаболизм соединений на данной изформе.
Схема 4.
эстрон нортриптилин хлорпромазин пулегон мексилетин
Данные РСА комплекса 1А2 изоформы с -нафтофлавоном (2hi4), взятые в Protein Data Bank, были использованы для докинга субстратов в ее полости. Исследовано расположение реакционных центров (атомов, участвующих в гидроксилировании и деалкилировании при метаболизме) субстратов относительно атома железа гема с целью прогноза направлений метаболических реакций, катализируемых изоформой цитохрома P450 1A2. Выделено 2 группы субстратов, в каждой из которых реакционные центры локализуются примерно в одном и том же месте пространства полости изоформы, это позволило предположить реализацию двух
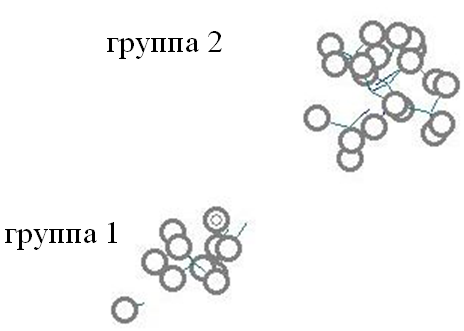 Рис. 25. Суперпозиция реакционных центров 14 субстратов изоформы 1A2. Выделены атомы водорода. Рис. 25. Суперпозиция реакционных центров 14 субстратов изоформы 1A2. Выделены атомы водорода. | различных механизмов метаболизма (Рис.25). В группу 1 попали ацетаминофен, хлорпромазин, эстрон, мексилетин, (R)-напроксен, (S)-напроксен, 9-цис-ретиналь, ретиналь. В группу 2 попали, амитриптилин, нортриптилин, фенацетин и т.д. Обнаружено, что, как правило, реакционные центры перечисленных молекул являются ближайшими к атому железа гема, располагающимися на расстоянии от 3.03 до 5.68, что допускает возможность |
взаимодействия этих атомов с координированным у железа гема кислородом в случае образования соответствующего кислородного комплекса. Исключение составляют молекулы хлорпромазина, эстрона, ретиналя. Для данных молекул, ближайшими к атому железа гема оказываются атомы, для которых экспериментального подтверждения метаболических реакций на изоформе 1А2 не было найдено в литературе, однако есть экспериментальные данные о протекании реакции по данным атомам при метаболизме на других изоформах, например, положение 8 хлорпромазина. Можно предположить, что на 1А2 изоформе 8-гидроксилирование хлорпромазина может протекать одновременно с 7-гидроксилированием. Расстояния от атома железа гема до указанных атомов представлены на Рис 26а. Аналогичная ситуация может наблюдаться для эстрона и ретиналя.
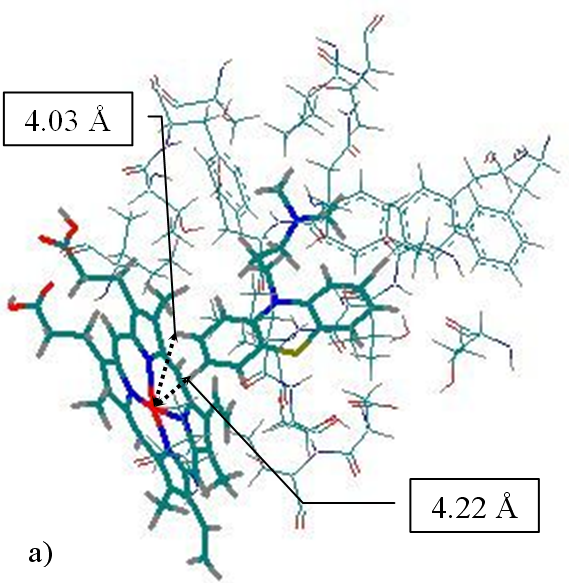
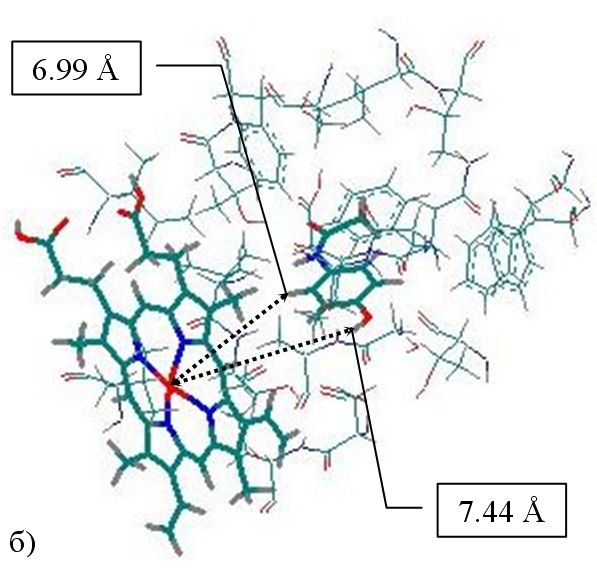
Рис. 26. Расположение молекул а) хлорпромазина и б) ацетаминофена в полости изоформы 1A2 по результатам моделирования. Показаны только те аминокислотные остатки, которые образуют сокращенные контакты с молекулами исследуемой выборки. Выделены молекулы субстратов и гем
В ряде случаев (например, ацетаминофен) реакционный центр субстратов расположен от атома железа гема на расстояниях, превышающих 7 (Рис.26б), что практически исключает возможность непосредственного взаимодействия атомов реакционного центра с кислородом, координированным у железа гема. В данных случаях можно предположить окисление по сетке водородных связей, образуемых с молекулами воды, либо участие аминокислотных остатков, образующих значительное количество сокращенных контактов. Подобное наблюдение согласуется с результатами моделирования комплексов субстратов с 3А4 изоформой. Аминокислотные остатки 1А2 изоформы, образующие сокращенные контакты с субстратами, представлены на Рис.27а. Показано, что подавляющее большинство таких контактов является липофильными. Рассмотрение комплексов с использованием алгоритма CoCon показало, что наилучшей является взаимосвязь, представленная выражением (16):
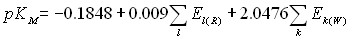
График полученной зависимости представлен на Рис. 27б. CrossR2=0,79, R=0,92, S= 0,09. Показано, что активными центрами лиганда, параметры взаимодействия с которыми определяют величину pKM, являются в основном атомы водорода, участвующие в метаболизме, а также атомы водорода метильных и метиленовых групп, определяющие периферию молекулы и ее гидрофобные взаимодействия с белком (Рис.28). Аналогичные исследования были проведены для субстратов другой наиболее распространенной изоформы 2С9, рассмотренной в п.п.3.8.
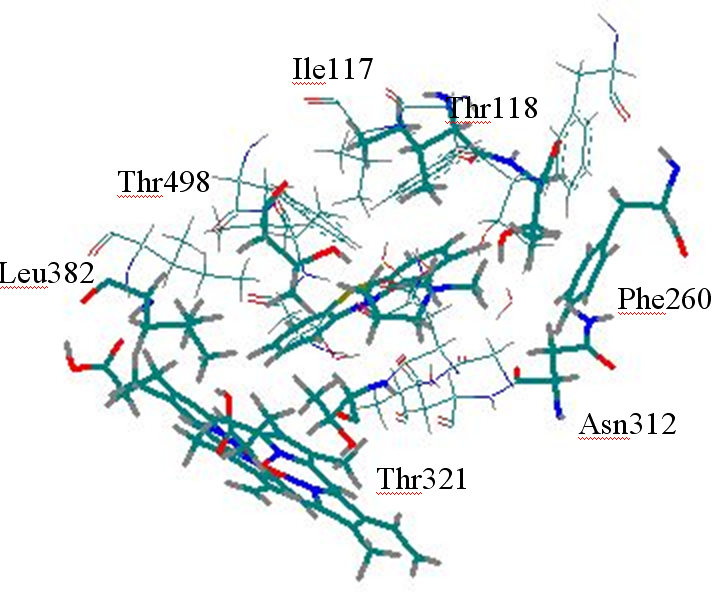
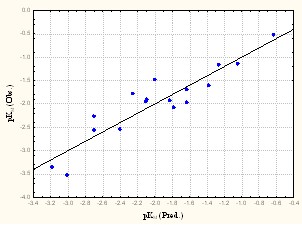
Рис. 27. Субстраты 1А2 изоформы: а) аминокислотные остатки, образующие сокращенные контакты с субстратами; б) расчетные (определенные с использованием алгоритма CoCon) и экспериментальные величины pKM субстратов
 Рис. 28. Активные центры лиганда – хлорпромазина, определенные CoCon в расчетных комплексах «1А2 изоформа - субстрат» Рис. 28. Активные центры лиганда – хлорпромазина, определенные CoCon в расчетных комплексах «1А2 изоформа - субстрат» | В главе 4 рассматривается дизайн новых перспективных соединений противоопухолевого, противовоспалительного, противовирусного и антидепрессантного действий, осуществленный на основании полученных в п.п.3.1-3.4 зависимостей. Всего было сконструировано около 150 активных структур. Для этого был применен алгоритм DesPot, выполняющий топологическую |
вариацию матриц смежности соединений с известной активностью.
Принципиально, данный подход заключается в обмене фрагментов активных структур для дизайна новой молекулы, активность которой прогнозируется с использованием решающих правил, поученных в рамках алгоритмов BiS/MC, ConGO, CoCon. Данный алгоритм представлен в работе [Потемкин В.А., Гришина М.А., Пожиленкова Г.В., Микушина К.М., Лауфер С. Генетический алгоритм молекулярного 3D дизайна биологически активных соединений// Биомедицинская химия.- 2004. Т. 50.- Прил. № 1.- С. 42 – 48]. Количественные модели, определенные в п.п.3.5-3.8, позволили провести прогноз метаболизма для найденных структур. Часть из них была синтезирована и прошла успешные биологические испытания in vivo. Данные соединения показаны на Рис.29 и 30.
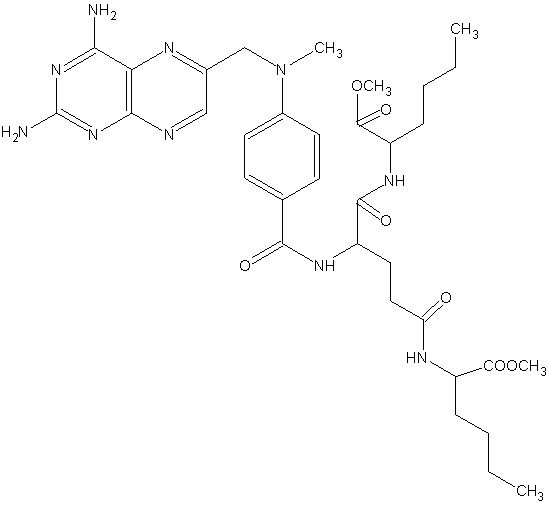
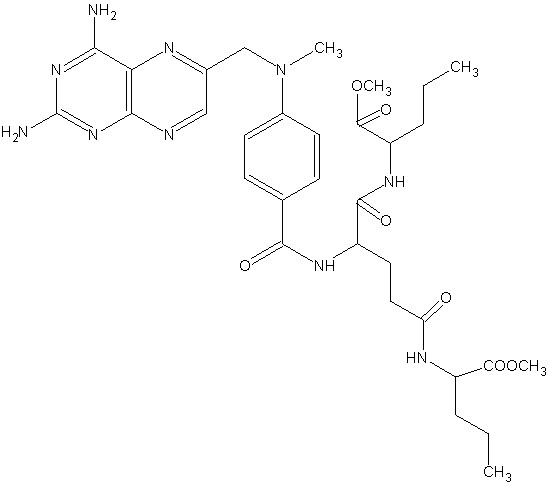
Рис. 29. Новые противоопухолевые средства: предсказанные на основании обучающих правил, представленных в работе, синтезированные и протестированные в Миланском университете (Италия).
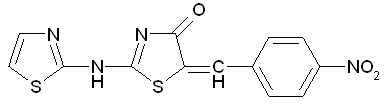
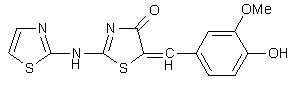
Рис. 30. Новые противовоспалительные средства: предсказанные на основании решающих правил, представленных в работе, синтезированные и протестированные в университете Аристотеля г. Салоники (Греция).
Выводы:
- Предложена комбинация компьютерных методов для анализа и прогноза биологически активных соединений, позволяющая
- производить детальное рассмотрение внешнего (характеристики поля) и внутреннего (параметры электронной структуры) строения биологически активных соединений с оценкой их таутомерно-конформационного состояния в момент взаимодействия с биологическим рецептором;
- осуществлять докинг соединений на сайте рецептора;
- определять количественные закономерности величины активности от параметров поля молекул, их электронной структуры, характеристик взаимодействия в модельных комплексах «рецептор-лаганд».
Данная комбинация включает алгоритм ProK для оценки таутомерно-конформационного равновесия в водных растворах органических соединений, 3D/4D QSAR алгоритмы BiS/MC и ConGO для анализа внешнего поля молекул, докинга соединений, рассмотрения параметров электронной структуры биологически активных соединений, алгоритм CoCon для анализа комплексов «рецептор-лиганд».
- Впервые в предложенной комбинации методов использован алгоритм AlteQ для оценки электронного строения молекул, с хорошим качеством воспроизводящий данные прецизионного РСА при значительно меньших временных затратах по сравнению с квантово-химическими подходами.
- С помощью данной комбинации компьютерных методов проведен анализ и выявлены детали механизмов биологического действия органических соединений: противоопухолевых средств – ингибиторов DHFR, противовоспалительных средств – ингибиторов p38 MAP киназы, противовирусных средств – ингибиторов нейраминидазы; изучено селективное действие антидепрессантных средств - ингибиторов 5HT1A-рецептора – в присутствии конкурирующего 1-AR рецептора.
- Данная комбинация методов использована для определения закономерностей в реакциях метаболизма субстратов на различных изоформах цитохрома Р450 (1А2, 3А4, 2С9 и др.).
- Для всех рассмотренных в работе соединений определены таутомерно-конформационные формы, встраивающиеся в рецептор, выделены фармакофорные и антифармакофорные фрагменты электронной структуры соединений.
- Выполнено построение комплексов «рецептор-лиганд». Сопоставление более 70 расчетных комплексов с данными РСА показало хорошее согласие между ними: найдено, что среднеквадратичное отклонение координат атомов молекул в расчетной и экспериментальной ориентациях в рецепторе, как правило, не превышает 1, что показывает более высокое качество прогноза строения комплексов «рецептор-лиганд» по сравнению с мировыми аналогами.
- Найдены количественные закономерности биологической активности от параметров внешнего поля молекул, их электронной структуры, характеристик взаимодействия в комплексах «рецептор-лиганд». Полученные количественные модели позволяют описать величины БА соединений (pIC50%, pGI50%, Ef, pKM и др.) с коэффициентами корреляции не менее 0.90.
- Полученные количественные закономерности использованы для конструирования новых лекарственных средств с оценкой возможности их метаболизма на изоформах цитохрома Р450. Четыре предсказанных соединения (два из них противоопухолевого и два противовоспалительного действий) синтезированы и прошли успешные предклинические биологические испытания in vivo в Миланском университете (Италия) и университете Аристотеля г. Салоники (Греция). Показано, что данные соединения обладают большей активностью по сравнению с используемыми в лечебной практике лекарственными средствами аналогичного терапевтического действия.
Основное содержание диссертации опубликовано в работах:
- Potemkin V. A., Bartashevich E. V., Grishina M. A., Guccione S. An Alternative Method for 3D-QSAR and the Alignment of Molecular Structures: BiS (Biological Substrate Search)// Rational Approaches to Drug Design. Proceedings of the 13th European Symp. on Quantitative Structure-Activity Relationships, QSAR 2000. Heinrich-Heine-Universitt, Dsseldorf, Germany, 27 August-1 September, 2000.- Barcelona: Prous Science Publishers, 2001.- P. 349 – 353.
- Потемкин В.А., Гришина М.А., Белик А.В., Чупахин О.Н. Исследование количественной взаимосвязи структура – антибактериальная активность производных хинолона// Хим.-фарм. журн.- 2002.- Т. 36.- № 1.- С. 22 – 25.
- Барташевич Е.В., Гришина М.А., Потемкин В.А., Белик А.В. Метод мультиконформационного моделирования пространственной формы молекулы// Журн. структ. химии.- 2002.- Т. 43.- № 6.- С. 1120 – 1127.
- Гришина М.А., Барташевич Е.В., Потемкин В.А., Белик А.В. Генетический алгоритм для прогноза строения и свойств молекулярных агломератов в органических веществах// Журн. структ. химии.- 2002.- Т. 43.- № 6.- С. 1128 – 1133.
- Потемкин В.А., Арсламбеков Р.М., Барташевич Е.В., Гришина М.А., Белик А.В., Перспикаче С., Гуччионе С. Мультиконформационный метод анализа биологической активности молекулярных структур// Журн. структ. химии.- 2002.- Т. 43.- № 6.- С. 1134 – 1138.
- Mikuchina K., Potemkin V., Grishina M., Laufer S. 3D QSAR analysis and pharmacophore modelling of p38 MAP kinase inhibitors using BiS algorithm// Arch. Pharm. Pharm. Med. Chem.- 2002.- Vol. 335.- N 1.- P. C74.
- Потемкин В.А., Гришина М.А., Федорова О.В., Русинов Г.Л., Овчинникова И.Г., Ишметова Р.И. Теоретическое исследование противотуберкулезной активности мембранотропных подандов// Хим.-фарм. журн.- 2003.- Т. 37.- № 9.- С. 17 – 21.
- Потемкин В.А., Гришина М.А., Пожиленкова Г.В., Микушина К.М., Лауфер С. Генетический алгоритм молекулярного 3D дизайна биологически активных соединений// Биомедицинская химия.- 2004. Т. 50.- Прил. № 1.- С. 42 – 48.
- Гришина М.А., Потемкин В.А., Микушина К.М., Пожиленкова Г.В., Лауфер С. Комбинированный подход к анализу биологической активности соединений// Биомедицинская химия.- 2004. Т. 50.- Прил. № 1.- С. 68 – 76.
- Погребной А.А., Рябинин В.Е., Барташевич Е.В., Гришина М.А., Потемкин В.А. Подход к отбору веществ с заданными метаболическими свойствами// Биомедицинская химия.- 2004. Т. 50.- Прил. № 1.- С. 77 – 84.
- Потемкин В.А., Гришина М.А., Барташевич Е.В., Зракова Т.Ю., Погребной А.А. Моделирование взаимодействий изоформы 2Е1 цитохрома Р450 человека с субстратами// Биофизика.- 2005.- Т. 50.- № 3.- С. 418 – 422.
- Гришина М.А., Погребной А.А., Потемкин В.А., Зракова Т.Ю. Теоретическое исследование субстратной специфичности изоформ цитохрома Р450// Хим.-фарм. журн.- 2005.- Т. 39.- № 10.- С. 3 – 7.
- Гришина М.А., Потемкин В.А., Барташевич Е.В., Синяев А.Н., Русинов Г.Л,, Латош Н.И., Ганебных И.Н., Корякова О.В., Ишметова Р.И. Моделирование комплексов производных 1,2,4,5-тетразина с органическими аминами// Журн. Структ. химии.- 2006.-47.- N 6.- с. 1165-1171.
- Потемкин В.А., Гришина М.А., Барташевич Е.В. Моделирование ориентации молекул лекарственных средств в полости рецептора в рамках алгоритма BiS// Журнал структурной химии. - 2007.-48.- N 1.- с. 153-158.
- Худина О.Г., Щегольков Е. В., Бургарт Я. В., Кодесс М. И., Салоутин В. И., Кажева О. Н., Шилов В. Г., Дьяченко О. А., Гришина М. А., Потемкин В. А., Чупахин О. Н. Исследование геометрической изомерии в ряду фторалкилсодержащих 2-арилгидразон-1,2,3-трионов // Журнал органической химии.- 2007.- Т. 43.- N 3.- С. 381-389
- Щур, И. В.; Худина, О. Г.; Бургарт, Я. В.; Салоутин, В. И.; Гришина, М. А.; Потемкин, В. А. Синтез, строение и комплексообразующая способность фторалкилсодержащих 2,2`-(4,4`-бифенилдигидразоно)-бис(1,3-дикарбонильных) соединений//Журнал органической химии. - 2007. - Т. 43.-N 12. - С. 1780-1786.
- Grishina M.A., Bartashevich E.V., Pereyaslavskaya E.S. and Potemkin V.A. A Novel Techniques for Virtual Discovery for Study of Multistage Bioprocesses// Drugs of the Future.- 2007.- Vol. 32, Suppl. A.- P. 27.
- Pereyaslavskaya E.S., Potemkin V.A., Grishina M.A., Bartashevich E.V. The analysis of pharmacophoric parts of DHFR – inhibitors using 3D-QSAR algorithm BiS/MC and X-ray data// Drugs of the Future.- 2007.- Vol. 32, Suppl. A.- P. 87.
- Grishina M.A., Potemkin V.A. A novel approach to pattern recognition for drugs// Drugs of the Future.- 2007.- Vol. 32, Suppl. A.- P. 112.
- Potemkin V. A novel technique for virtual discovery for study of multistage bioprocesses// 234th ACS National Meeting.- Boston, August 19-23, 2007.- Book of Abstr.- P. COMP253.
- Гришина М.А., Потемкин В.А., Матерн А.И. Теоретическое исследование реакций окисления акриданов// Журн. структ. химии.- 2008.- Т. 49.- № 1.- С. 13 – 18.
- Potemkin V.A., Grishina M.A. A new paradigm for pattern recognition of drugs. Journal of Computer Aided Molecular Design. 2008.- 22.- P. 489-505.
- Гришина М.А., Потемкин В.А. GonGO N 2007614593 зарегистрировано в Реестре программ для ЭВМ 31 октября 2007 г., заявка N 2007613592 от 7 сентября 2007 г.
- Гришина М.А., Потемкин В.А. ProK N2008610190, зарегистрировано в Реестре программ для ЭВМ 9 января 2008 г., заявка N 2007613591 от 7 сентября 2007 г.
- Гришина М.А., Потемкин В.А. BiS/MC (multi-conformational), зарегистрировано в Реестре программ для ЭВМ 18 февраля 2008 г., заявка N 2007613594 от 7 сентября 2007 г.
- Гришина М.А., Потемкин В.А., Погребной А.А., Ившина Н.Н. Исследование конформационных состояний субстратов изоформы 3A4 цитохрома P450 // Биофизика. - 2008. - Т.53. - Вып.5. - С.758-765.
- Переяславская Е.С., Потемкин В.А., Барташевич Е.В., Гришина М.А., Русинов Г.Л., Федорова О.В., Жидовинова М.С., Овчинникова И.Г. Теоретическое исследование туберкулостатической активности соединений дигидропиримидинового ряда// Хим.-фарм. журн.- 2008.- Т. 42.- № 11.- С. 23 – 26.
- Potemkin V., Grishina M. Princiles for 3D/4D QSAR>
- Багрянская И.Ю., Гришина М.А., Cафина Л.Ю., Селиванова Г.А., Потемкин В.А., Гатилов Ю.В. Рентгеноструктурные и квантово-топологические исследования межмолекулярных взаимодействий в кристаллах частично фторированных хинолинов// Журн. структ. химии.- 2008.- Т. 49.- № 5.- С. 933 – 942.
- Potemkin V, Pogrebnoy A., Grishina M. A Technique for Energy Decomposition in the Study of "Receptor-Ligand" Complexes //Journal of Chemical Information and Modeling - 2009.- Vol. 49.- № 6.- P. 1389 – 1406
- Афонькина Е.С., Гришина М.А., Ившина Н.Н., Матвеев Г.А., Потемкин В.А. Реализация параллельной версии программы расчёта лекарственных средств с использованием Т-Системы с открытой архитектурой (OpenTS)// Труды международной конференции "Программные системы: теория и приложения", ИПС РАН им. А.К. Айламазяна, г. Переславль-Залесский, май 2009/ Под редакцией С.М. Абрамова и С.В. Знаменского.- Т. 1.- Переславль-Залесский: Изд-во "Университет города Переславля", 2009.- С. 217 – 224. ISBN 978-5-901795-16-3.
- Kuzmicheva G.A., Jayanna P.K., Eroshkin A.M., Grishina M.A., Pereyaslavskaya E.S., Potemkin V.A., Petrenko V.A. Mutations in fd phage major coat protein modulate affinity of the displayed peptide// Protein Engineering Design and Selection.- 2009.-Vol.22.-P.631-639.
- Potemkin V., Galimova O., Grishina M. Cinderella’s Shoe for virtual drug discovery screening and design// Drugs of the Future. – 2010.- Vol. 35.-P. 14–15.
- Grishina M., Potemkin V. Electron-nuclear quintessence of drugs therapeutic action. Drugs of the Future. 2010.- Vol. 35. – P. 130 – 131.
- Afonkina E., Palko N., Toreeva N., Potemkin V., Grishina M. Restricted docking of the DHFR inhibitors to the DHFR receptor. Drugs of the Future. 2010. Vol. 35. P. 182.
- Афонькина Е.С. Потемкин В.А., Гришина М.А. Теоретическое исследование реакции несимметричных 3,6-дизамещённых сим-тетразинов с некоторыми енаминами// Химия гетероциклических соединений.- 2010.-Т. 518.- N 8. - P. 1217 – 1222.
- Гришина М.А., Потемкин В.А., Погребной А.А., Сысаков Д.А. Моделирование комплексов субстратов с цитохромом P450 2C9. -2010. - Т. 44.- N 5.- с. 16-20.
- Sushko I., Novotarskyi S., Krner R., Pandey A.K., Rupp M., Teetz W., Brandmaier S., Abdelaziz A., Prokopenko V.V., Tanchuk V.Y., Todeschini R., Varnek A., Marcou G., Ertl P., Potemkin V., Grishina M., Gasteiger J., Schwab C., Baskin I.I., Palyulin V.A., Radchenko E.V., Welsh W.J., Kholodovych V., Chekmarev D., Cherkasov A., Aires-de-Sousa J., Zhang Q-Y., Bender A., Nigsch F., Patiny L., Williams A., Tkachenko V., Tetko I.V. Online chemical modeling environment (OCHEM): web platform for data storage, model development and publishing of chemical information// J. Comput. Aided Mol. Des.- 2011.- Vol. 25. - P. 533-554.
- Гришина М.А., Матвеев Г.А., Потемкин В.А. BiS_p N 2010615221 зарегистрировано в Реестре программ для ЭВМ 13 августа 2010 г.
- Потемкин В.А., Матвеев Г.А., Гришина М.А. CiS_p N 2010615222 зарегистрировано в Реестре программ для ЭВМ 13 августа 2010 г.
[1] Название соответствует идентификатору в Protein Data Bank