

Горь викторович синтез и свойства биологически активных соединений, содержащих no-донорный фрагмент
На правах рукописи
Серков Игорь Викторович
СИНТЕЗ И СВОЙСТВА БИОЛОГИЧЕСКИ АКТИВНЫХ СОЕДИНЕНИЙ, СОДЕРЖАЩИХ NO-ДОНОРНЫЙ ФРАГМЕНТ
02.00.10 – Биоорганическая химия
Автореферат диссертации на соискание ученой степени
доктора химических наук
Черноголовка, 2010
Работа выполнена в Учреждении Российской академии наук
Институте физиологически активных веществ РАН
Официальные оппоненты:
| доктор химических наук, член-корреспондент РАН, профессор, директор Института биохимической физики им. Н.М. Эмануэля РАН | Варфоломеев Сергей Дмитриевич |
| доктор химических наук, Институт биоорганической химии им. М.М. Шемякина и Ю.А. Овчинникова РАН | Формановский Андрей Альфредович |
| доктор химических наук, Институт элементорганических соединений им. А.Н. Несмеянова РАН | Кочетков Константин Александрович |
Научный консультант:
| доктор химических наук, профессор | Безуглов Владимир Виленович |
Ведущая организация: Учреждение Российской академии наук
Институт проблем химической физики РАН
Защита состоится «15» февраля 2011 г. в 14 часов на заседании
специализированного совета Д 002.102.01 при Учреждении Российской академии наук Институте физиологически активных веществ РАН по адресу: 142432, Московская обл., Ногинский р-н, г. Черноголовка, Северный проезд, 1
С диссертацией можно ознакомиться в библиотеке Учреждения Российской академии наук Института физиологически активных веществ РАН
Автореферат разослан «…» ноября 2010 г.
Ученый секретарь специализированного совета
кандидат химических наук Великохатько Т.Н.
ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТы
Актуальность проблемы. Одно из направлений современной медицинской химии – концепция «многофункциональных лекарств». В рамках этой парадигмы понятию лекарственного средства как «магической пули» с максимальной селективностью, действующего на одну, строго определённую мишень, противопоставлена широта фармакологического действия и способность лекарственного вещества взаимодействовать с несколькими мишенями. Многофункциональные лекарства содержат в своей структуре несколько фармакофоров, действие которых дополняет друг друга. Такие препараты имеют более предсказуемый фармакокинетический профиль, у них существенно снижен риск несовместимости с другими препаратами за счёт уменьшения количества прописываемых пациенту лекарств. Одним из направлений в создании таких полифункциональных соединений является введение в молекулу известного лекарственного препарата фрагмента, являющегося генератором оксида азота (NO). NO – химически активное соединение, которое непрерывно продуцируется в организме из аминокислоты аргинина с помощью NO-синтаз. NO является внутри- и межклеточным мессенджером со многими важными биохимическими и физиологическими свойствами. Эта маленькая молекула не только передает биохимические сигналы, влияя таким образом на различные биологические системы, включая центральную нервную, сердечно-сосудистую и иммунную системы, но и действует как важный регулятор основных клеточных процессов. Нарушение биосинтеза и метаболизма NO приводит к тяжелым заболеваниям, таким как инфаркт миокарда, ишемическая болезнь сердца, астма, нейродегенеративные заболевания (болезнь Альцгеймера, болезнь Паркинсона), диабет и многие другие. Добавление к молекуле лекарственного средства NO-донорного фрагмента придает «старому» препарату новые свойства за счет активации физиологических механизмов, активируемых NO, и предотвращения различные патологий, вызванных отсутствием или недостатком генерации NO. Такая модификация существенно изменяет фармакологические свойства лекарственных веществ, расширяя области их применения и снижая, в большинстве случаев, присущие немодифицированным соединениям побочные эффекты. Это увеличивает фармакологический потенциал и эффективность лекарственного препарата.
Таким образом, развитие методов создания гибридных препаратов содержащих NO-донорный фрагмент, разработка способов введения NO-генерирующей группировки в молекулу биологически активного соединения – актуальные задачи в плане создания новых высокоэффективных лекарственных препаратов. Эти направления вносят существенный вклад в познание химических свойств биологически активных соединений и в теорию дизайна потенциальных лекарственных препаратов.
Данная работа является частью плановой тематики Института физиологически активных веществ РАН и выполнялась в соответствии с общесоюзной программой АН СССР «Простагландины», государственной научно-технической программой «Атеросклероз». Поддержана грантами: РФФИ 94-03-09326-а «Разработка новых способов синтеза эфиров и тиоэфиров природных простагландинов», 00-04-48797-а «Исследование молекулярного механизма и нейрорецепторной активности эндогенных каннабиноидов и их нитроэфиров как новых эффективных биорегуляторов и потенциальных нейрокорректоров», 02-04-22002-НЦНИ-а (PICS 1582) «Изучение отношений структура-активность в ряду новых производных полиненасыщенных жирных кислот как потенциальных нейропротекторов», 04-04-49515-а «Эфиры полиненасыщенных жирных кислот. Синтез и исследование их влияния на ионные каналы в мембране нервных клеток».
Цель и задачи работы. Основная цель – разработка подходов и способов создания гибридных физиологически активных соединений, содержащих NO-донорный фрагмент, на основе биологически активных спиртов как основы потенциальных полифункциональных высокоэффективных лекарственных препаратов в рамках фундаментальной проблемы биоорганической химии – установление связи между структурой биологических соединений и их физиологической активностью. В задачи исследования входили: разработка общих способов введения NO-донорной группировки на основе нитратов спиртов в молекулы биологически активных соединений; синтез таких модифицированных соединений на основе различных классов фармакологически значимых агентов; изучение фармакологических свойств синтезированных соединений.
Научная новизна. Разработаны новые конструкции и способы создания гибридных многофункциональных физиологически активных соединений на основе нитратов биологически активных спиртов – NO-донорных фармакофоров. Разработаны способы синтеза эфиров и амидов простагландинов и полиненасыщенных жирных кислот с нитратами спиртов и аминоспиртов. Показана универсальность разработанных способов введения NO-донорных группировок на основе нитратов биологически активных спиртов. Впервые синтезированы нитраты природных гидроксиаминокислот и пептиды на их основе. Впервые описано нитрование аллильной гидроксильной группы в молекуле простагландина. Проведено исследование биологических свойств синтезированных гибридных соединений и изучено влияние NO-генерирующего фрагмента на их физиологическую активность. Таким образом, создано новое направление в конструировании новых многофункциональных лекарственных препаратов.
Практическая значимость работы. На основе предложенных методов получения гибридных соединений, содержащих нитраты биологически активных спиртов как NO-донорный фрагмент, синтезированы 1,3-динитроглицерино-вые и нитроэтиленгликолевые эфиры, а также амиды с нитроаминоспиртами ряда физиологически активных соединений. В качестве исходных фармакофоров были использованы простагландины, полиненасыщенные жирные кислоты, нестероидные противовоспалительные средства (индометацин, ибупрофен, ацетилсалициловая кислота), цефалоспорин G. Это означает, что разработанные методы применимы к разнообразным классам биологически активных соединений и могут использоваться при создании гибридных препаратов и с другими фармакофорами. Разработаны препаративные способы синтеза нитроксисерина и нитрокситреонина. Проведено нитрование аллильной гид–роксильной группы в молекуле простагландина и синтезированы 15-нитра–ты простагландинов. Предложено использование триметилсилильной защит–ной группы для синтеза фторангидридов простагландинов. Показано, что введение NO-донорной группировки в молекулу фармакофора резко меняет фармакологические свойства последнего, что открывает путь к направленному конструированию новых лекарственных препаратов. Так, созданная на основе 1,3-динитроглицеринового эфира простагландина Е1 мазь оказалась эффективной при лечении ожоговых травм у экспериментальных животных.
Автор защищает созданное новое направление в конструировании прототипов новых лекарственных препаратов на основе многофункциональных физиологически активных соединений, содержащих нитраты биологически активных спиртов – NO-донорных фармакофоров, и способы его реализации.
Апробация работы. Основные результаты диссертации были представлены на: 9-ой Международной конференции «Простагландины и родственные соединения» (Флоренция, 1994 г.), ХIV Международном симпозиуме по медицинской химии (Маастрихт, 1996 г.), Международной научно-практической конференции «Биологически активные вещества и новые продукты в косметике» (Москва, 1996 г.), IV Съезде Белорусского общественного объединения фотобиологов и биофизиков «Молекулярно-клеточные основы функционирования биосистем» (Минск, 2000 г.), 11-ой Международной конференции «Простагландины и лейкотриены: фундаментальная наука и новое клиническое применение» (Флоренция, 2000 г.), I Международной конференции «Химия и биологическая активность азотистых гетероциклов и алкалоидов» (Москва, 2001 г.), Международном симпозиуме «Успехи в синтетической, комбинаторной и медицинской химии» (Москва, 2004 г.), XVIII Менделеевском съезде по общей и прикладной химии (Москва, 2007 г.), III Съезде фармакологов России (Санкт-Петербург, 2007 г.), Научной конференции «Органическая химия для медицины» (Черноголовка, 2008 г.), VII Всерос-сийской научной конференции «Химия и медицина, ОРХИМЕД–2009» (Уфа, 2009 г.), IV Российском симпозиуме «Белки и пептиды» (Казань, 2009), VIII Всероссийской конференции «Химия и медицина» (Уфа, 2010), 5-ой Международной конференции «Биологические основы индивидуальной чувствительности к психотропным средствам» (Москва, 2010).
Публикации. Основные результаты исследований, проведенных по теме диссертации, изложены в 15 статьях, 15 тезисах докладов и описаниях к 7 патентам и авторским свидетельствам.
Объем диссертации и ее структура. Диссертация изложена на 201 странице машинописного текста, содержит 17 схем, 9 таблиц, 14 рисунков и состоит из введения, 3 глав, выводов и списка цитируемой литературы объемом 329 ссылок. В первой главе «Обзор литературы» обсуждены методы синтеза многофункциональных соединений, содержащих органические нитраты как прототипов гибридных лекарственных препаратов нового поколения. Во второй главе описаны разработанные автором способы создания гибридных многофункциональных физиологически активных соединений на основе нитратов биологически активных спиртов и приведены данные по исследованию биологических свойств синтезированных гибридных соединений и влиянию NO-донорного фрагмента на их физиологическую активность. В третьей главе содержатся экспериментальные данные.
ОСНОВНОЕ СОДЕРЖАНИЕ РАБОТы
- Концепция гибридных лекарственных препаратов,
содержащих NO-донорный фрагмент.
В связи с актуальной в последнее время концепцией «одно лекарство – много мишеней» нами разработана концепция новых гибридных лекарственных соединений, т.е. соединений, объединяющих в одной молекуле несколько фармакофоров. В качестве первого фармакофора выступает биологически активное соединение (как природное вещество, так и молекула действующего начала известного лекарственного препарата), а второй фрагмент представляет собой связанную окись азота (NO), которая является важным внутри- и межклеточным регулятором многих основных биохимических процессов и оказывает влияние на различные системы организма, включая центральную нервную, сердечно-сосудистую и иммунную системы. В результате вовлечения в механизм действия гибридного препарата эффектов высвобождаемой окиси азота может повыситься эффективность исходного фармакофора и измениться его фармакологический профиль, в том числе – произойти уменьшение побочных эффектов, присущих немодифицированному соединению. Все это существенно расширяет область возможного применения данных гибридных препаратов. В качестве NO-донорного фармакофора нами предложены органические нитраты спиртов, и этот выбор был сделан сознательно по следующим причинам. Во-первых, у органических нитратов отсутствует спонтанная генерация NO, что позволяет направленно доставлять такие соединения в клетки-мишени без потери NO-генерирующей активности. Во-вторых, выделение NO из нитратов спиртов – это тиолзависимый процесс, требующий участия специфических белков, что может обеспечить генерацию NO в нужное время и в нужном месте. В-третьих, при генерации NO из органических нитратов высвобождается гидроксильная группа, восстанавливая структуру исходного фармакофора. Образующееся природное или известное лекарственное соединение подвергается метаболизму по стандартным путям, и в организме не происходит накопление «неприродных» фрагментов с неизученными свойствами. И, наконец, в-четвертых, превращение в нитроэфиры меняет фармакокинетику и лигандные свойства исходного фармакофора. Например, соединение становится более липофильным и может легче проникать через мембраны клеток или гематоэнцефалический барьер.
Для введения нитратной группы в молекулу фармакологически активного вещества нами было использовано два подхода. Первый подход основан на прямом нитровании гидроксильных групп с образованием так называемых «безлинкерных» гибридных соединений. В случае отсутствия в молекуле гидроксильной группы или необходимости ее сохранения в конечном гиб–ридном соединении, а также при лабильности исходного соединения в условиях нитрования NO-донорный фрагмент присоединяли с помощью дополнительной группировки. Этот подход приводит к линкерным гибридным соединениям, когда два фармакофора соединены с помощью третьего дополнительного компонента. Принцип построения этих гибридных соединений показан на рис. 1.
Рис. 1
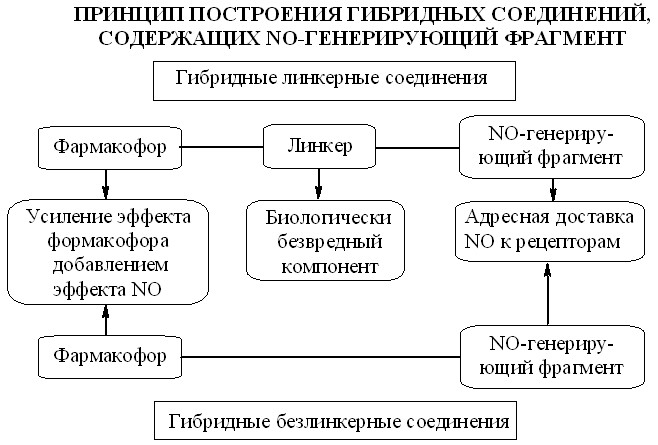
В качестве линкеров для присоединения NO-донорного фрагмента мы использовали природные биологически активные спирты (глицерин, этилен-гликоль) и аминоспирты (этаноламин, 3-амино-1,2-пропандиол и другие). Такие природные линкеры были выбраны не случайно, а для того, чтобы в процессе метаболизма сконструированного гибридного соединения в организме не происходило образования чужеродных фрагментов, создающих дополнительную нагрузку на метаболический аппарат организма. Из спиртов были синтезированы соответствующие нитраты, которые вводили в молекулу в качестве NO-донорного фрагмента.
1.2. Общие подходы к синтезу гибридных соединений,
содержащих NO-донорный фрагмент
Наиболее удобным и распространенным способом синтеза нитроксисоединений остается прямое нитрование гидроксильной группы азотной кислотой или ее смесями, если при этом не происходит побочных процессов, связанных с деструкцией молекулы исходного спирта. Этим способом нами синтезированы из аминоспиртов линкеры, содержащие NO-донорный фрагмент (NO-линкеры), а также безлинкерные гибридные соединения, содержащие NO-генерирующую нитроксигруппу на основе простагландинов, насыщенных и ненасыщенных жирных кислот и гидроксиаминокислот.
Используя полученные нитраты аминоспиртов, а также 1,3-динитрат глицерина и мононитроэтиленгликоль в качестве NO-линкеров, нами разработаны способы синтеза гибридных соединения на основе простагландинов, полиненасыщенных жирных кислот, цефалоспорина G и ряда нестероидных противовоспалительных препаратов (ацетилсалициловая кислота, аспирин, индометацин, ибупрофен). Все эти соединения имеют карбоксильную группу, что позволило присоединить NO-линкеры с помощью эфирной или амид-ной связи.
2. Синтез биологически активных соединений, содержащих
NO-донорный фрагмент.
2.1. Синтез линкеров, содержащих нитроксигруппы в качестве
NO- донорного фрагмента (NO-линкеров).
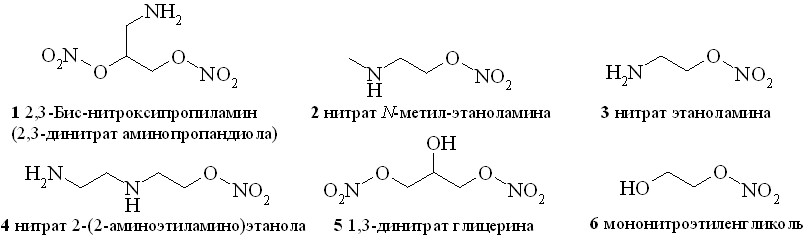
В качестве NO-линкеров использованы глицерин, этиленгликоль и различные аминоспирты. Из них сначала были получены соответствующие нитроксипроизводные – нитраты аминоспиртов (1–4), 1,3-динитрат глицерина (5) и мононитроэтиленгликоль (6) (рис. 2). Нитраты аминоспиртов (1–4) получены нитрованием соответствующих аминоспиртов в виде азотнокислых солей. В качестве нитрующего агента был использован раствор 100% азотной кислоты в хлористом метилене. Выделившуюся в ходе реакции воду связывали с помощью рассчитанного количества уксусного ангидрида. В результате нерастворимые в хлористом метилене азотнокислые соли нитроксиаминоспиртов выпадали в осадок и легко отделялись от реакционной массы с помощью простого фильтрования.
Рис. 2
2.2. Нитраты гидроксиаминокислот.
Природные гидроксиаминокислоты серин и треонин являются не только распространенными компонентами белков и пептидов, но и обладают соб-ственной биологической активностью. В литературе описаны синтезы нитроэфиров дипептидов, содержащих серин и треонин. Однако данные дипептиды содержали защитную Cbz-группировку на аминогруппе. Удалить эту защитную группировку после нитрования для получения незащищенного нитроксидипептида с сохранением нитратной группы невозможно.
Главная трудность синтеза нитратов серина и треонина заключается не в самом химическом процессе нитрования, а в выделении целевых соединений из реакционной смеси. Серин и треонин очень хорошо растворимы в воде и плохо в органических растворителях. Поэтому их невозможно выделить из реакционной смеси с помощью экстракции даже после превращения в нитроксисоединения. Использование временных защитных группировок по амино- или карбоксильной группе для повышения липофильности получаемых аминокислот также не приводит к желаемому результату. Во-первых, такие защитные группы должны обладать взаимоисключающими свойствами: быть устойчивыми в кислых условиях реакции нитрования и удаляться также в кислых условиях. Использовать же защитные группировки, удаляемые в щелочных условиях или в реакциях гидрогенолиза, невозможно из-за лабильности нитроэфирной группировки в этих условиях. Во-вторых, даже если бы удалось подобрать соответствующую защитную группу, опять встала бы проблема выделения целевого продукта, но уже после удаления защиты.
Наиболее удачной нитрующей смесью в синтезе нитратов L-серина (7), D-серина (8) и L-треонина (9) (рис. 3) оказался раствор 100% азотной кислоты в хлористом метилене, ранее использованный для получения нитратов аминоспиртов (1–4).
Рис. 3
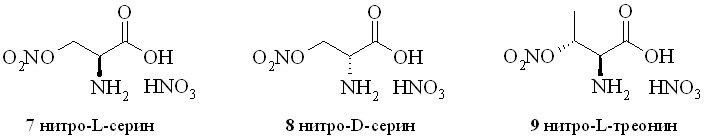
Следует отметить, что этаноламин и его аналоги достаточно хорошо растворимы в хлористом метилене, что позволило нам использовать гомогенную реакционную смесь с медленным прибавлением раствора аминоспирта к нитрующей смеси. В данном случае исходные аминокислоты можно было растворить только в воде. Однако это привело бы к нежелательному разбавлению азотной кислоты с неизбежным снижением её нитрующей способности, а также к расслоению водной и органической фаз. Поэтому мы прибавляяли к нитрующему раствору сухую аминокислоту. Образующаяся в ходе реакции вода растворяла азотнокислые соли нитратов гидроксиаминокислот. По окончании выделившуюся в ходе реакции воду связывали добавлением расчетного количества уксусного ангидрида, целевые соединения выпадали в виде кристаллического осадка и отделялись простым фильтрованием. Следует отметить, что добавление уксусного ангидрида вначале реакции нитрования приводит к образованию нежелательных ацетатов, трудноотделимых от целевых нитратов.
С целью изучения химических свойств полученных нитратов серина и треонина на их основе нами были синтезированы дипептиды – N-Boc-(L)-пролин-(D)-нитросерин (10), N-Cbz-(D,L)-пролин-(D)-нитросерин (11), N-Boc-глицин-(L)-нитротреонин (12) и N-Boc-(D)-аланин-(D)-нитросерин (13), а также защищенные нитроаминокислоты – N-Boc-(D)-нитросерин (14) и N-Fmoc-(L)-нитротреонин (15) (рис. 4).
Рис. 4
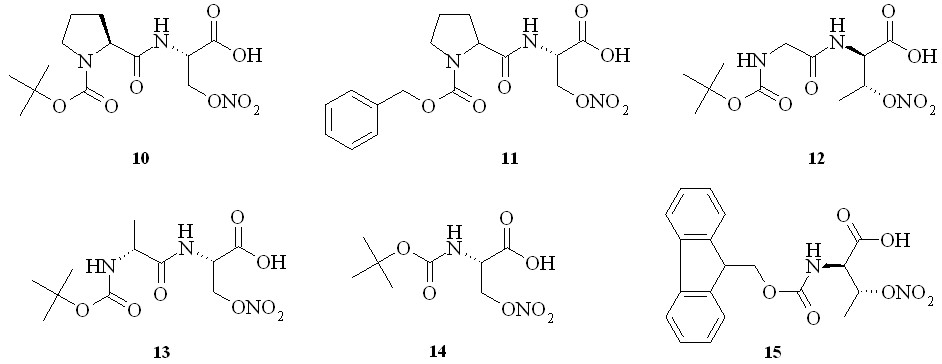
Дипептид N-Boc-(L)-пролин-(D)-нитросерин (10) был получен конденсацией N-Boc-(L)-пролина (16) с (D)-нитросерином (8) с использованием «хлорформатного» метода через смешанный ангидрид (17), который затем без выделения конденсировали с (D)-нитросерином (8) в дипептид (10) (схема 1). Аналогично из N-Boc-глицина и (L)-нитротреонина (9) получили дипептид (12), а из N-Cbz-(D,L)-пролина и N-Boc-(D)-аланина и (D)-нитросерина (8) – дипептиды (11) и (13).
Схема 1
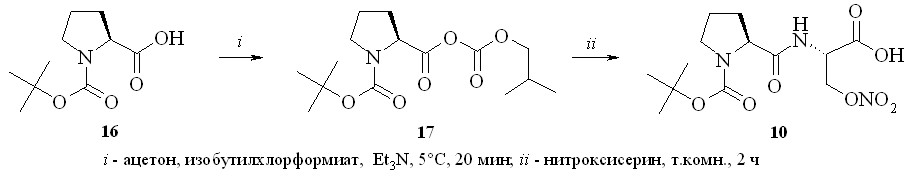
Защищенные по аминогруппе нитроаминокислоты получали по стандартным методикам синтеза таких производных, а именно с использованием ди-трет-бутил-дикарбоната при получении соединения (14) и флуоренилметилхлорформата – для соединения (15).
2.3. Безлинкерные соединения, содержащие NO-донорный фрагмент,
на основе простагландинов и ненасыщенных жирных кислот.
Простагландины (ПГ) являются полифункциональными соединениями, для которых характерно проявление широкого спектра физиологической активности, что препятствует широкому применению природных ПГ в качестве лекарственных препаратов из-за множественных побочных эффектов. Биологические эффекты NO и ПГ близки по своей физиологической направленности. Поэтому создание гибридных NO-генерирующих соединений на основе ПГ представляется весьма перспективным для разработки более эффективных и безвредных препаратов. Нами синтезированы как линкерные, так и безлинкерные представители таких простагландиновых производных.
Представителями безлинкерных соединений на основе ПГ являются 15-нитраты 11-дезокси-ПГE1 (20) и его метилового эфира (21) (схема 2). Нами впервые синтезированы 15-нитроксипроизводные простагландинов прямым нитрованием гидроксильной группы 11-дезокси-ПГЕ1 (18) или его метилового эфира (19) (см. таблицу 1).
Схема 2
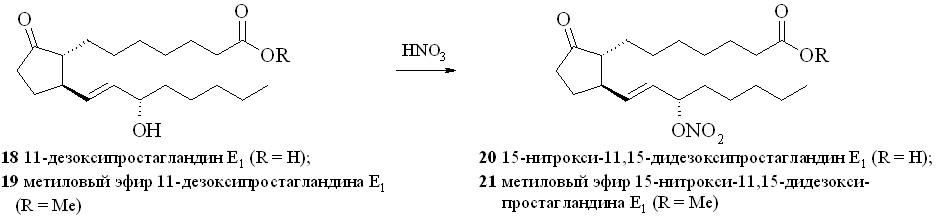
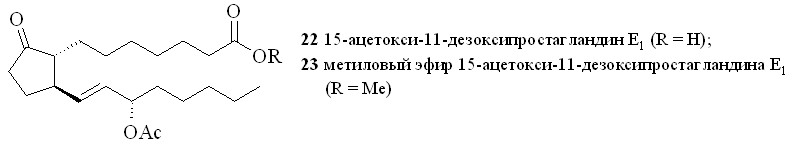
Применение стандартной смеси, состоящей из 67% азотной кислоты и уксусного ангидрида, для нитрования гидроксильной группы в ПГ (18, 19) приводило к образованию целевых нитратов (20, 21). Однако выход этих нитратов был невелик и не превышал 35%, при этом образовывалось неожиданно много 15-ацетоксипроизводных (22) и (23). Исключение уксусного ангидрида из реакционной среды позволило избежать образования ацетатов. При незначительном снижении выхода целевых нитратов конверсия простагландина в этом случае составила 80–85% за счёт возврата непрореагировавшего ПГ с помощью хроматографии. Замена уксусного ангидрида на концентрированную серную кислоту практически не влияла на выход нитратов, а вместо ацетатов образовывались трудно идентифицируемые продукты дегидратации (табл. 1). Растворитель, в котором проводилось нитрование, практически не влиял на выход и чистоту целевого нитрата. Наилучший результат по чистоте и выходу 15-нитратов достигнут при проведении реакции нитрования концентрированной (более 96%) HNO3 в неполярном растворителе. При нитровании небольших количеств ПГ азотную кислоту получали in situ из нитрата натрия или калия и серной кислоты. При использовании различных нитрующих смесей и способов нитрования выходы нитратов свободной кислоты (20) и метилового эфира (21) были практически одинаковы. Однако следует отметить, что метиловый эфир (21) устойчив при хранении, тогда как кислота (20) в отсутствие растворителей довольно быстро подвергается разложению с потерей молекулы азотной кислоты.
Таблица 1. Условия нитрования и выходы 15-нитрокси-11,15-дидезоксипростагландинов Е1.
| Условия реакции | Выход нитрокси- производного, % | Побочные продукты |
| NaNO3, H2SO4, CH2Cl2, 20°С, 1 ч | 6575 | практически отсутствуют |
| HNO3 (100%), CH2Cl2, 10°С, 40 мин | 7585 | практически отсутствуют |
| HNO3 (67%), диоксан, 20°С, 4 ч | 1525 | исходный ПГ |
| HNO3 (67%), Ас2О, диоксан, 20°С, 4 ч | 2234 | 15-ОАс |
| HNO3 (67%), H2SO4, диоксан, 20°С, 4 ч | 2530 | продукты дегидратации |
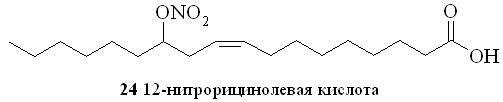
Прямым нитрованием азотной кислотой гидроксильной группы было синтезировано и безлинкерное соединение на основе гидроксипроизводного ненасыщенной жирной кислоты – 12-нитрорицинолевая кислота (24).
Рис. 5
2.4. Гибридные соединения, содержащие NO-донорный фрагмент,
на основе нестероидных противовоспалительных препаратов.
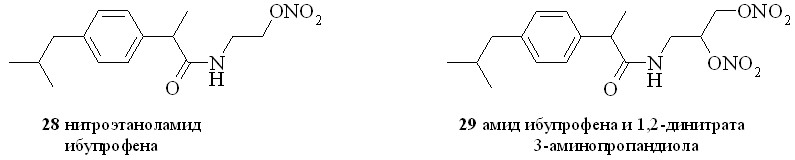
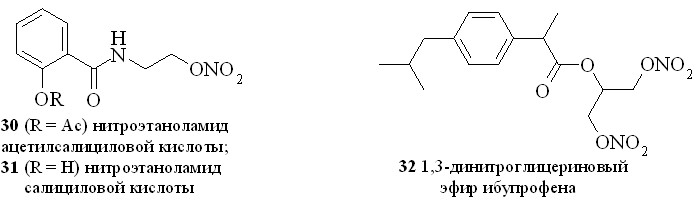
Нестероидные противовоспалительные препараты (NSAID) (аспирин, индометацин, напраксен, ибупрофен и др.) в течение многих лет применяются в качестве жаропонижающих и противовоспалительных средств. Присоединение к структуре NSAID фрагмента, генерирующего NO, позволило создать новый класс противовоспалительных препаратов, так называемых NO-генерирующих нестероидных противовоспалительных препаратов (NO-NSAID). Такие гибридные соединения, сохраняя противовоспалительную активность исходного препарата, обладают гораздо меньшим отрицательным воздействием на желудочно-кишечный тракт. Предполагается, что NO-NSAID защищают желудочно-кишечный тракт путем локального выделения NO, приводящего к усилению кровообращения в слизистой оболочке. Кроме того, NO сам обладает гастропротекторными свойствами. Нами синтезирован ряд NO-генерирующих NSAID, содержащих в качестве NO-донора нитроксигруппу, присоединенную к основной молекуле через аминоспирт в качестве линкера (2531), а также 1,3-ДНГ-эфиры ибупрофена (32) и индометацина (33) (рис. 6).
Рис. 6
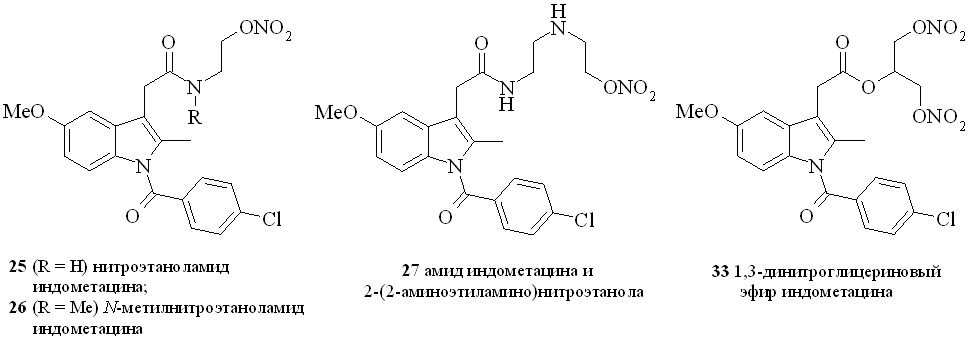
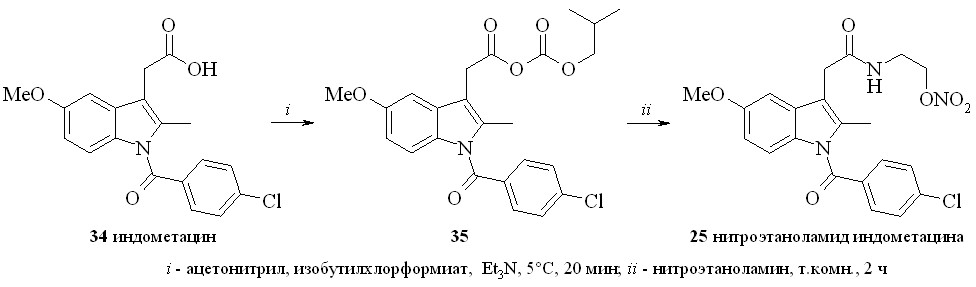
Амиды индометацина (25–27) синтезировали по методу «смешанных ангидридов» реакцией с изобутилхлорформиатом с последующей конденсацией с нитратами аминоспиртов (схема 3). Выход целевых соединений составлял 65–85%.
Схема 3
Использование этого метода для синтеза производных ибупрофена и салициловой кислоты не привело к желаемому результату. Реакция амидирования либо проходила с выходом не более 40%, либо основным продуктом реакции был изобутиловый эфир кислоты. Не дало положительных результатов и использование высокореакционноспособных имидазолидов. Наиболее удачным оказался способ синтеза через промежуточные хлорангидриды (схема 4).
Схема 4
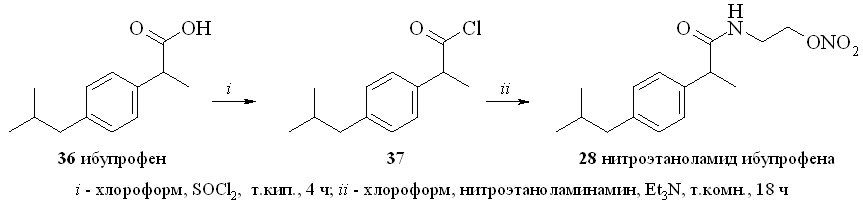
Сначала из ибупрофена (36) кипячением с SOCl2 в хлороформе синтезировали хлорангидрид (37). Затем реакционную смесь упаривали, получившийся хлорангидрид конденсировали с нитроэтаноламином (3) с образованием нитроэтаноламида ибупрофена (28). Аналогично были синтезированы амиды (2931). 1,3-ДНГ-эфиры ибупрофена (32) и индометацина (33) синтезированы по методу «смешанных ангидридов» с арилсульфокислотами, а эфир (32) – также через хлорангидрид (37).
2.5. NO-донорные производные цефалоспорина G.
Цефалоспорины широко применяются в медицине и по структуре и ме-ханизму действия близки к пенициллинам. Цефалоспорин G также использу-ется для ферментативного получения дезацетоксицефалоспорановой кислоты и синтеза разнообразных пролекарств. В последнем случае присоединенный к 3’-углеродному атому цефалоспорина фрагмент молекулы лекарственного соединения высвобождается в организме за счет ферментативной реакции. Значительно меньше внимания уделяется производным цефалоспоринов по карбоксильной группе. В качестве прототипа нового класса антибиотиков, содержащих NO-донорный фрагмент, был выбран 1,3-динитроглицериновый эфир цефалоспорина G (40), который является альтернативой синтезировано-го ранее конъюгата цефалоспорина с 3-морфолиносидноимином. Его синтезировали по методу «смешанных ангидридов» с арилсульфокислотами, в данном случае с 2,4,6-триизопропилбензолсульфокислотой (схема 5).
Схема 5
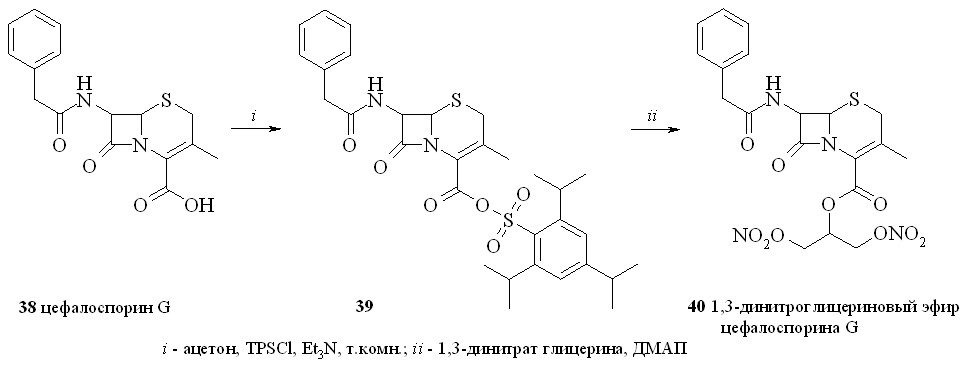
Сначала реакцией цефалоспорина G (38) с TPSCl синтезировали смешанный ангидрид (39), который затем в присутствии ДМАП конденсировали с 1,3-динитратом глицерина (5) с образованием 1,3-ДНГ-эфир цефалоспорина G (40).
2.6. Синтез гибридных линкерных соединений на основе ПГ и ПНЖК.
Для синтеза гибридных соединений на основе ПГ и полиненасыщенных жирных кислот (ПНЖК) с помощью NO-линкера применены два подхода. В первом случае использовали нитроаминоспирты (14), а также 1,3-динитрат глицерина (5) и мононитроэтиленгликоль (6), которые присоединяли к основной молекуле в виде сложноэфирной или амидной группировки, то есть присоединяли NO-линкеры. Во втором случае присоединяли только линкеры (аминоспирты или этиленгликоль), а затем проводили нитрование полученного амида или эфира.
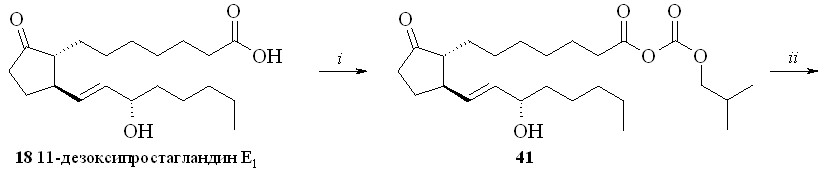
2.6.1. Амиды ПГ и ПНЖК с нитратами аминоспиртов.
Амиды ПГ с нитратами аминоспиртов получали методом «смешанных ангидридов» по стандартной методике (схема 6). Для этого ПГ, например 11-дезокси-ПГE1 (18), превращали в смешанный ангидрид (41), который без вы-деления вводили в реакцию с нитроэтаноламином (3) с образованием нитро-этаноламида 11-дезокси-ПГE1 (42).
Схема 6
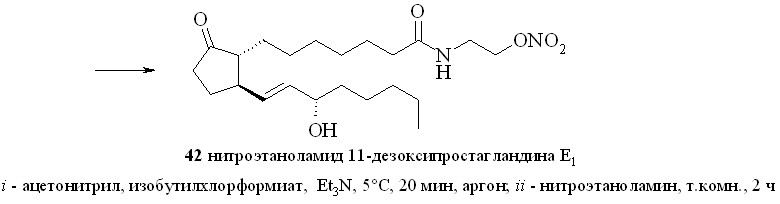
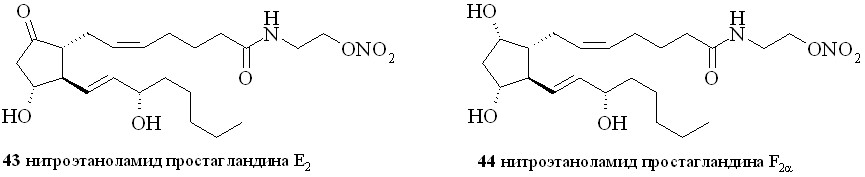
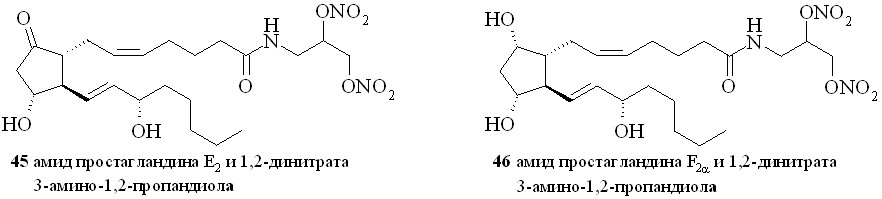
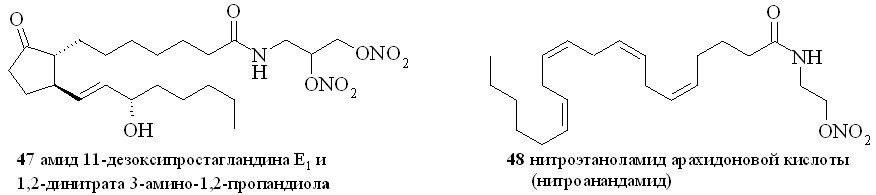
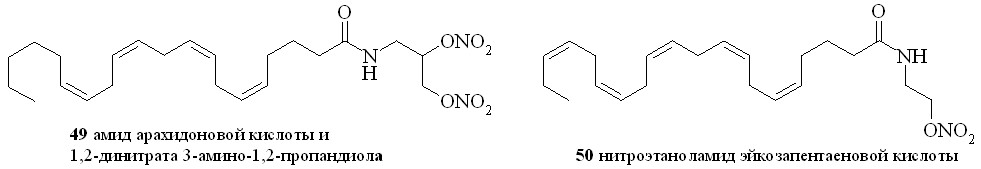
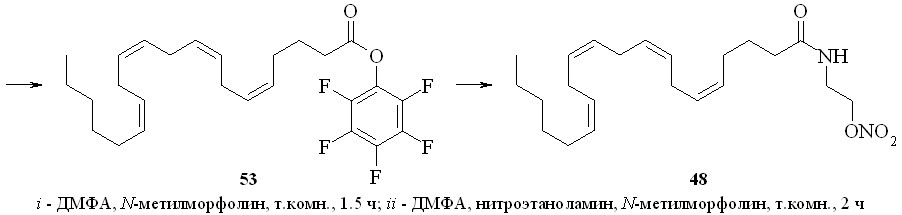
Аналогично синтезированы нитроэтаноламиды простагландинов Е2 (43), F2 (44), арахидоновой (нитроанандамид) (48) и эйкозапентаеновой (50) кислот, а также амиды ПГE2, ПГF2, 11-дезокси-ПГE1 и арахидоновой кислоты с 1,2-динитратом 3-амино-1,2-пропандиола (45, 46, 47 и 49) (рис. 7).
Рис. 7
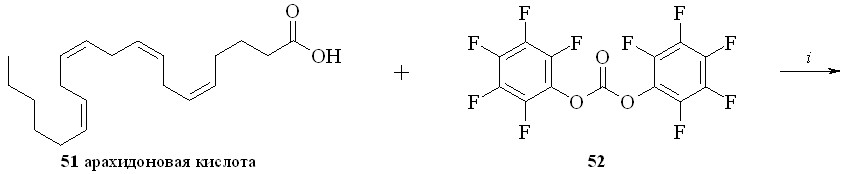
Для синтеза амидов жирных кислот с нитратами аминоспиртов был также использован метод активации карбоксильной группы через образование высоко реакционноспособного пентафторфенилового эфира (схема 7). По этому методу, например, из арахидоновой кислоты (51) реакцией с бис-пентафторфенилкарбонатом (52) получили пентафторфениловый эфир (53), который далее реакцией с нитроэтаноламином (3) превратили в нитроанандамид (48). Преимущество данного способа активации карбоксильной группы заключается в возможности предварительной наработки относительно устойчивого пентафторфенилового эфира жирной кислоты.
Схема 7
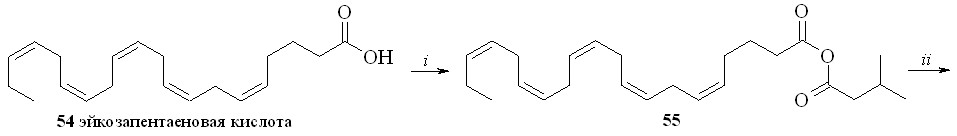
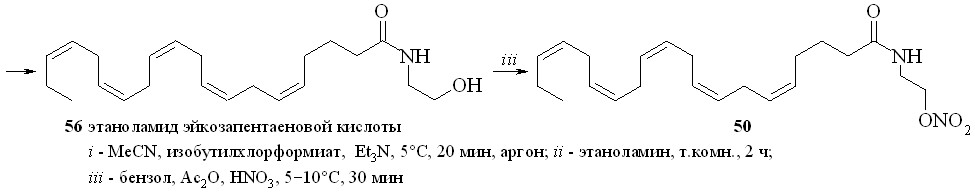
Нитроанандамид (48) и нитроэтаноламид эйкозапентаеновой кислоты (50) были синтезированы также вторым способом (схема 8), а именно нитрованием соответствующих этаноламидов. Так, из эйкозапентаеновой кислоты (54) и этаноламина через промежуточный смешанный ангидрид (55) был синтезирован этаноламид эйкозапентаеновой кислоты (56), из которого реакцией нитрования получали нитроэтаноламид эйкозапентаеновой кислоты (50). Преимущество этого метода заключается в том, что по ходу синтеза получаются этаноламиды ПНЖК, которые можно использовать как соединения сравнения при проведении биологических испытаний.
Схема 8
2.6.2. Эфиры ПГ и ПНЖК с нитратами спиртов.
При создании гибридных соединений кроме нитроксиаминов, присоединяемых через амидную связь, нами использованы «эфирные» NO-линкеры: 1,3-динитрат глицерина (5) и мононитроэтиленгликоль (6). Модификация карбоксильной группы ПГ путем превращения ее в эфиры с простыми или сложными спиртами часто используется для создания производных ПГ с целью модификации фармакологического профиля последних. В каждом конкретном случае для получения таких производных используются специально разработанные способы, так как универсальные способы синтеза сложных эфиров ПГ практически отсутствуют. Большинство методов синтеза эфиров карбоновых кислот основывается на активации карбоксильной группы с последующей реакцией образовавшегося активированного производного с соответствующим спиртом. Специфической проблемой в синтезах производных по карбоксильной группе таких полифункциональных соединений, как ПГ, является необходимость сохранения остальных функциональных групп (гидрокси- и кетогруппы) при активации карбоксильной группы.
2.6.2.1. Синтез 1,3-динитроглицериновых эфиров ПГ.
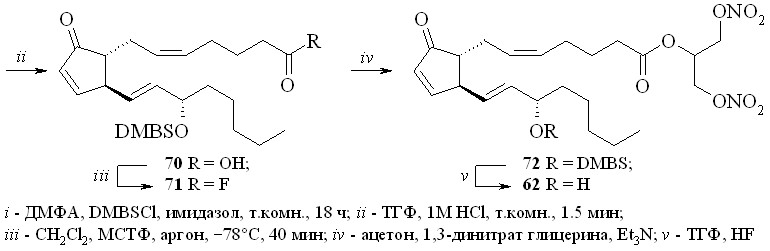
1,3-Динитроглицериновые эфиры (1,3-ДНГ-эфиры) ПГ были получены этерификацией природных ПГ 1,3-динитратом глицерина (5) через активацию карбоксильной группы исходного ПГ. Разработано и исследовано несколько способов такой этерификации: 1 – через смешанные ангидриды с арилсульфокислотами; 2 – через активированные ацилимидазолиды; 3 – путём реакции ПГ с 1,3-динитратом глицеринхлорформиата с последующей перегруппировкой в искомый эфир; 4 – через превращение ПГ в высоко реакционноспособный фторангидрид.
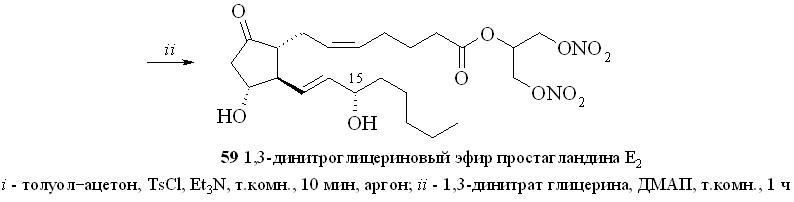
1. Арилсульфохлориды достаточно давно применяются как конденсирующие агенты в реакциях этерификации карбоновых и аминокислот, а также для образования фосфоэфирной связи в нуклеотидном синтезе. В зависимости от силы карбоновой кислоты и нуклеофильности спирта реакция этерификации протекает либо через образование промежуточного смешанного ангидрида, как в случае бензойной кислоты, либо через арилсульфонат спирта, как было постулировано для этерификации свободных аминокислот. В случае реакции ПГ с арилсульфохлоридами реакция, по-видимому, проходит через образование смешанного ангидрида ПГ и арилсульфокислоты. Так, все наши попытки получить этиловый эфир ПГ реакцией переэтерификации этилового эфира р-толуолсульфокислоты не привели к целевому соединению, а ТСХ-анализ продуктов реакции не выявил образования симметричного ангидрида ПГ. Из этого можно сделать вывод, что сначала происходит образование смешанного ангидрида ПГ с арилсульфокислотой, который затем подвергается нуклеофильной атаке спиртом с образованием сложного эфира ПГ. По этому методу сначала для предотвращения возможных реакций арилсульфохлорида с 1,3-динитратом глицерина простагландин, в данном случае ПГЕ2 (57), превращали в смешанный ангидрид (58) реакцией с арилсульфохлори-дом (р-толуолсульфохлоридом (TsCl) или 2,4,6-триизопропилбензолсульфо-хлоридом (TPSCl)) в присутствии триэтиламина (схема 9). После завершения данной стадии прибавляли 1,3-динитрат глицерина (5) и каталитическое количество диметиламинопиридина (ДМАП). В результате получали целевой 1,3-ДНГ-эфир ПГЕ2 (59).
Схема 9
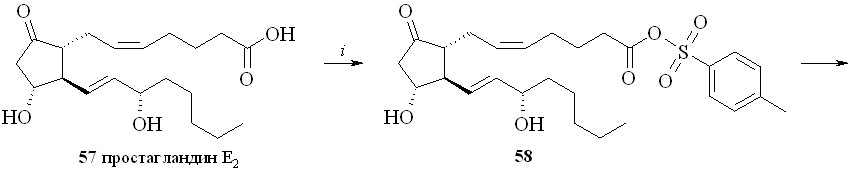
Недостаток этого метода – побочная реакция хлорирования аллильного гидроксила в положении 15 молекулы ПГ, особенно при использовании TsCl, что приводит к загрязнению целевого соединения трудноотделяемыми примесями и к уменьшению выхода эфира. Попытки избежать нежелательной реакции хлорирования заменой хлорангидридов арилсульфокислот на их активированные амиды – р-толуолсульфонилимидазол, триизопропилсульфонилимидазол, триизопропилсульфонилтриазол – оказались неудачными.
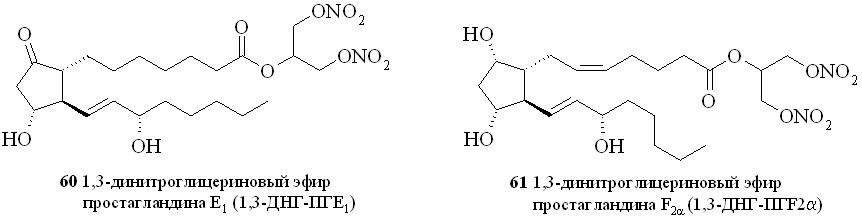
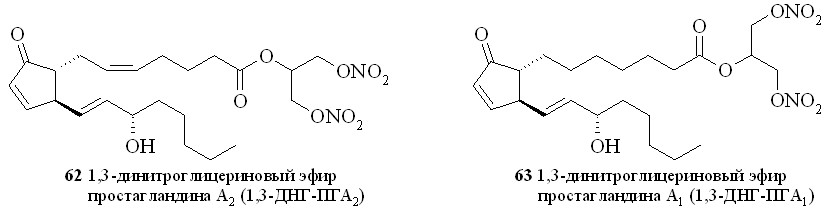
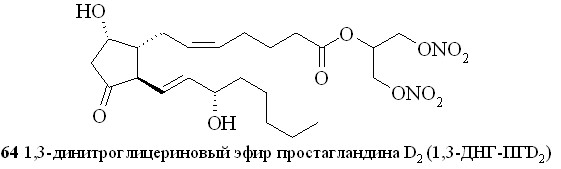
Этим методом помимо эфира 59 были синтезированы 1,3-ДНГ-эфиры простагландинов Е1 (60), F2 (61), A2 (62), A1 (63) и D2 (64) (рис. 8).
Рис. 8
2. Высоко реакционноспособные имидазолиды широко применяются для синтеза амидов и сложных эфиров кислот. В случае получения сложных эфиров необходимы кислотные катализаторы. Этот метод был применен нами для синтеза 1,3-ДНГ-эфиров ПГ (схема 10).
Схема 10

Сначала реакцией ПГЕ2 (57) с 1,1'-карбонилдиимидазолом (CDI) в ацетонитриле получали имидазолид (65), который без выделения конденсировали с 1,3-динитратом глицерина (5) в присутствии гидрохлорида пиридина и получали эфир ПГ. Однако выход целевого эфира (59) был невысок (3045%), что, по-видимому, связано с низкой реакционной способностью 1,3-динитрата глицерина (5).
3. Смешанные ангидриды с производными угольной кислоты часто используются для активации карбоксильной группы при получении амидов кислот. Кроме того, такие смешанные ангидриды под действием ДМАП могут претерпевать перегруппировку в сложный эфир и поэтому иногда применяются для получения труднодоступных эфиров. Это свойство смешанных ангидридов использовано нами для получения 1,3-ДНГ-эфиров ПГ (схема 11).
Схема 11
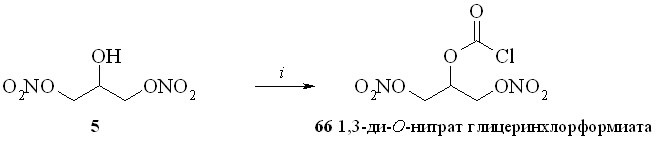
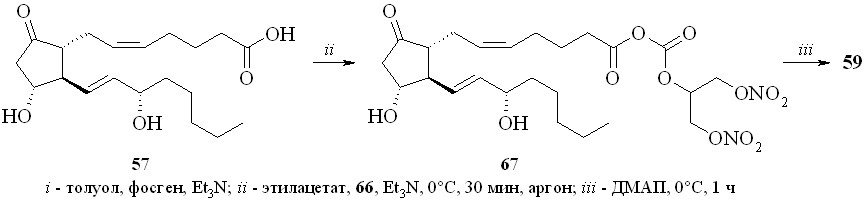
Сначала из фосгена и 1,3-динитрата глицерина (5) был синтезирован 1,3-динитрат глицеринхлорформиата (66) – достаточно устойчивое соединение, которое может быть дополнительно очищено перегонкой в вакууме. Реакцией хлорформиата (66) с ПГЕ2 (57) получили смешанный ангидрид (67), который не выделяли. К реакционной массе прибавляли ДМАП, что приводило к перегруппировке смешанного ангидрида в целевой 1,3-ДНГ-эфир (59). При использовании свежеприготовленного хлорформиата (66) выход реакции этерификации близок к количественному.
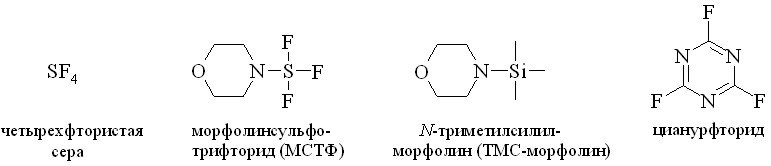
4. В практике для синтеза сложных эфиров широко применяются галоидангидриды, в частности хлорангидриды кислот. Однако ПГ наряду с карбоксильной группой содержат в своей структуре дополнительные гидроксильные группы, вовлекаемые в побочные реакции при получении хлорангидридов. Из-за этого галоидангидриды ПГ не привлекли внимание химиков как активированные производные для синтеза эфиров простагландинов. Нами при изучении реакции фторирования простагландинов было найдено, что превращение ПГ в его фторангидрид можно провести с помощью четырёхфтористой серы (SF4) и фторирующих агентов на её основе, в частности морфолиносульфотрифторида (МСТФ) (рис. 9), в мягких условиях. Полученный фторированием ПГА2 в качестве промежуточного соединения фторангидрид 15-фтор-15-дезокси-ПГА2 был превращен гидролизом в слабощелочной среде в 15-фтор-15-дезокси-ПГА2 и использован при синтезе 15-фтор-15-дезокси-ПГЕ2 в виде свободной кислоты.
Рис. 9
Однако, как было сказано выше, молекула ПГ помимо карбоксильной группы содержит одну или две гидроксильные группы, которые также подвергаются реакции фторирования аминотрифторсульфуранами. Поэтому для использования этого метода в синтезе производных по карбоксильной группе природных ПГ нами разработаны схемы синтезов фторангидридов, не затрагивающие гидроксильные группы молекулы. Для защиты гидроксильных групп применили временную их защиту силильными группировками, такими как трет-бутилдиметилсилильная (BDMS) и триметилсилильная (TMS), которые удаляли после получения соответствующих производных по карбоксильной группе ПГ.
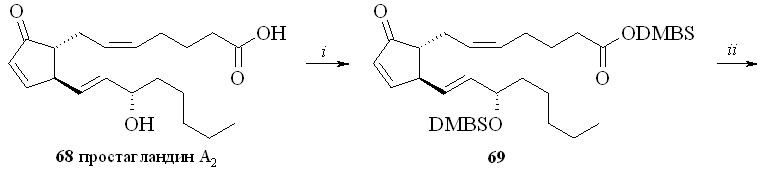
При использовании DMBS-защитной группировки простагландин, например ПГА2 (68), исчерпывающе силилировали трет-бутилдиметилхлорсиланом (DMBSCl) в присутствии имидазола (схема 12). Полученный DMBS-эфир 15-DMBS-ПГА2 (69) обрабатывали раствором 30%-ной Н2О2 в метаноле. В этих условиях защитная силильная группировка удаляется только с карбоксильной группы. Аналогичный результат получается, если вместо раствора перекиси водорода в метаноле использовать водный раствор 1М соляной кислоты в ТГФ. Эта реакция проходит значительно быстрее (примерно за 1 минуту), в то время как при использовании перекиси водорода для завершения реакции требуется около часа. Однако при использовании соляной кислоты из-за быстроты процесса затруднён контроль протекания реакции гидролиза, и при небольшом удлинении времени реакции происходит частичное деблокирование гидроксильной группы. Полученный 15-DMBS-ПГА2 со свободной карбоксильной группой (70) фторировали МСТФ с образованием фторангидрида (71) при сохранении силильного эфира на гидроксильной группе. Затем реакцией нуклеофильного замещения с 1,3-динитратом глицерина (5) в присутствии триэтиламина синтезировали эфир (72). Силильную защитную группировку с гидроксильной группы удаляли кислым гидролизом и получали 1,3-ДНГ-эфир ПГА2 (62).
Схема 12
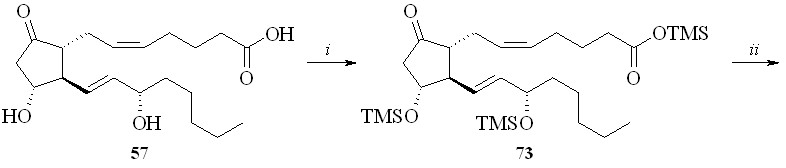
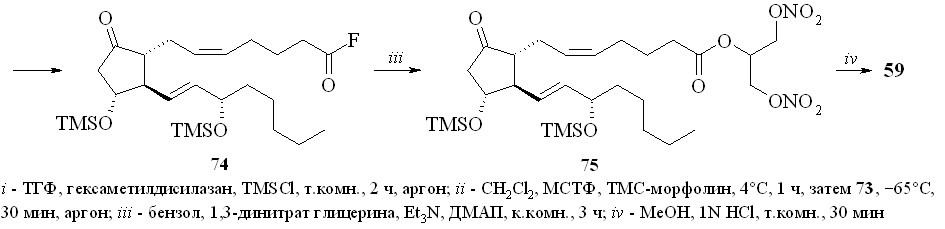
Недостатком этого метода является его многостадийность и, главное, необходимость проведения хроматографической очистки полученных промежуточных силилированных простаноидов. Более предпочтительной была бы такая силильная группа, которая удалялась в ходе реакции фторирования только с карбоксильной группы и сохранялась бы на гидроксильных группах. Наше внимание привлекла триметилсилильная защитная группировка. Сложные триметилсилиловые эфиры неустойчивы в условиях реакции фторирования MSTF, однако при этом происходит также деблокирование и гидроксильных групп. После серии экспериментов нами было найдено, что если в качестве фторирующего агента использовать не сам MSTF, а его смесь с N-триметилсилилморфолином (TMS-морфолин) в соотношении 1:1, то защитная TMS-группировка на гидроксильных группах сохраняется, в то время как на карбоксильной она замещается на фтор с образованием фторангидрида. Со-гласно приведенной схеме, раствор ПГЕ2 (57) в ТГФ обрабатывали смесью гексаметилдисилазана и триметилхлорсилана (схема 13). Полученное TMS-производное ПГЕ2 (73) фторировали эквимолярной смесью MSTF и TMS-морфолина. Полученный фторангидрид (74) реакцией с 1,3-динитратом глицерина (5) в присутствии триэтиламина превращали в эфир (75) и после кислого гидролиза TMS-эфира получали целевой 1,3-ДНГ-эфир ПГЕ2 (59).
Схема 13
Использование BDMS- или TMS-защитной группировки зависит от структуры конечного соединения. Как показали наши исследования, при по-лучении производных по карбоксильной группе природных ПГ предпочтение следует отдать использованию TMS-защитной группировки. Для синтеза же карбоксипроизводных фтордезоксианалогов ПГ лучше подходит BDMS-за-щита. В этом случае одновременно с удалением BDMS-защитной группиров-ки с карбоксильной группы можно частично деблокировать и гидроксильные группы (в основном аллильную гидроксильную группу при С-15 атоме ПГ), что не удаётся сделать в случае TMS-эфиров. Затем в реакции фторирования моносилилированного простаноида действием МСТФ происходит образование фторангидрида при одновременном фторировании и гидроксильной группы. Этим методом из ПГЕ2 (57) был получен 1,3-ДНГ-эфир 15-фтор-15-дезокси-ПГЕ2 (80) (схема 14). ПГЕ2 исчерпывающе силилировали с образованием производного (76). Полученный силилированный ПГ (76) обрабаты
Схема 14
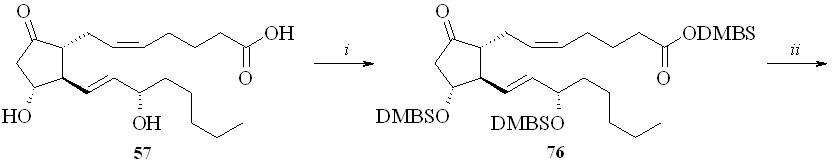
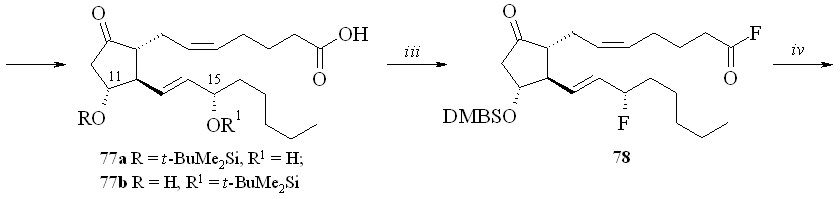
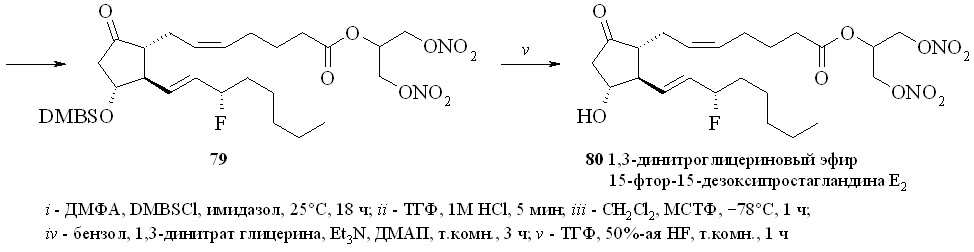
вали раствором 1М соляной кислоты в ТГФ в течение 5 минут. При этом удаляется силильная защита с карбоксильной группы, а также с одной из гидроксильных групп и образуется смесь моносилильных производных ПГЕ2 – 11-DMBS-ПГЕ2 (77а) и 15-DMBS-ПГЕ2 (77b) (с преобладанием изомера со свободной гидроксильной группой в положении 15), которую разделяли хроматографически. Выделенный таким образом 11-DMBS-ПГЕ2 (77а) фторировали MSTF. Синтезированный фторангидрид силилированного 15-фтор-15-дезокси-ПГЕ2 (78) конденсировали с 1,3-динитратом глицерина (5) и получали эфир (79). Защитную силильную группировку удаляли с помощью плавиковой кислоты и получали 1,3-ДНГ-эфир 15-фтор-15-дезокси-ПГЕ2 (80). В качестве фторирующего агента при синтезе фторангидридов можно также использовать цианурфторид, который реагирует исключительно с карбоксильной группой, не затрагивая при этом гидроксильные группы. Однако эта реакция проходит в присутствии пиридина в качестве основания, отчего при синтезе фторангидридов этим методом частично происходит катализируемая пиридином реакция получившегося фторангидрида со свободными гидроксильными группами молекулы того же ПГ, что приводит к смеси трудно идентифицируемых продуктов внутри- и межмолекулярной конденсации и, как следствие, к резкому снижению выхода целевого соединения. Однако цианурфторид оказался удобным реагентом при синтезе фторангидридов ПНЖК, не имеющих в своей структуре гидроксильных групп, и был использован нами для синтеза их производных (см. ниже).
Сравнение приведённых способов синтеза 1,3-ДНГ-эфиров простагландинов показывает, что наилучшие результаты достигаются при использовании метода, основанного на реакции перегруппировки смешанного ангидрида простагландина и 1,3-динитрата глицеринхлорформиата (66) (метод 3). Выход целевого соединения при этом способе конденсации приближается к максимальному (~90–95%), особенно при использовании свежеполученного хлорформиата. Однако получение соответствующего хлорформиата требует применения высокотоксичного фосгена. Хороший выход (около 75%) достигнут при использовании в качестве промежуточных соединений высокореакционных фторангидридов ПГ (метод 4). Тем не менее, данный способ требует предварительного получения TMS-эфиров ПГ с последующей процедурой удаления силильной защиты. Наиболее практичным способом синтеза 1,3-ДНГ-эфиров ПГ оказался метод с использованием в качестве промежуточных соединений смешанных ангидридов с арилсульфокислотами (метод 1). Этот способ позволяет получать 1,3-ДНГ-эфиры с неплохим выходом (65–70%), без дополнительных процедур и использования опасных реагентов. Наименее удачным оказался метод активации карбоксильной группы через имидазолиды (метод 2). Выход в этом случае составлял не более 35–40%, что, по-видимому, связано с особенностью 1,3-динитрата глицерина.
2.6.2.2. 1,3-Динитроглицериновые и нитроэтиленгликолевые эфиры ЖК.
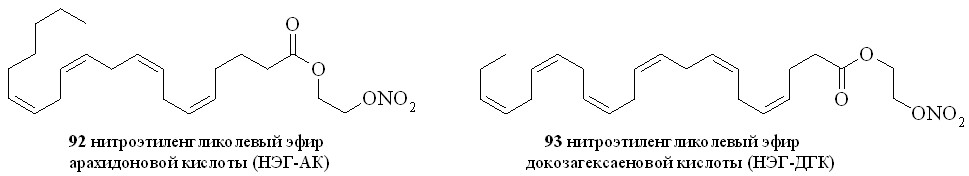
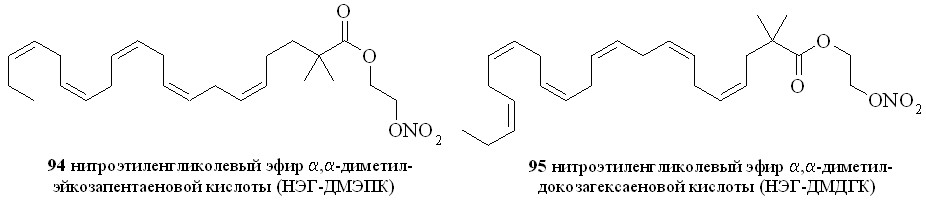
Используя разработанные методы синтеза, мы получили также 1,3-ДНГ-эфиры ряда жирных кислот – арахидоновой (81), докозагексаеновой (82), эйкозапентаеновой (83),,-диметиларахидоновой (84), линолевой (85), линоленовой (86), пальмитиновой (87), каприловой (88) и лауриновой (89) (рис 10), а также мононитроэтиленгликолевые эфиры арахидоновой (90), докозагексаеновой (91), эйкозапентаеновой (92),,-диметиларахидоновой кислот (93),,-диметилэйкозапентаеновой кислот (94) и,-диметилдокозагексаеновой кислот (95) (рис 11).
Рис. 10. 1,3-Динитроглицериновые эфиры жирных кислот
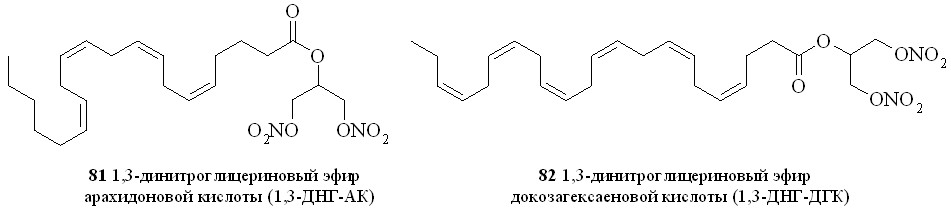
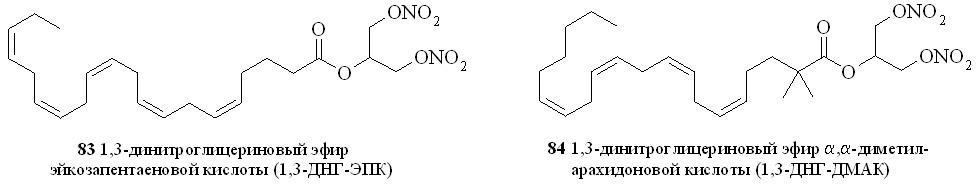
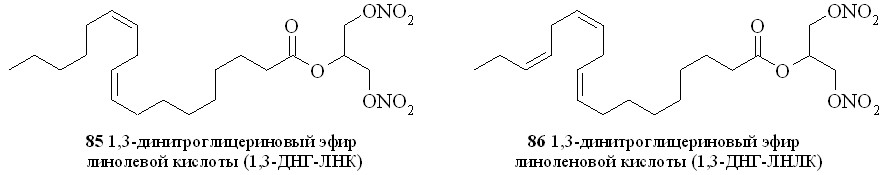
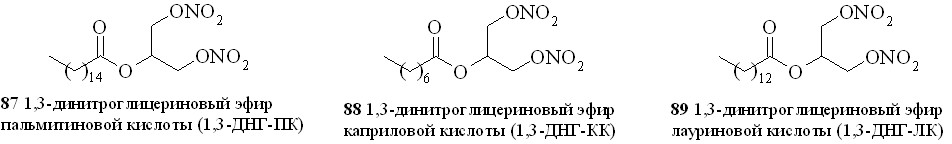
Рис. 11. Нитроэтиленгликолевые эфиры полиненасыщенных жирных кислот

Представленные жирные кислоты не имеют в своей структуре гидроксильных групп. Поэтому при синтезе их 1,3-ДНГ и НЭГ эфиров нами был широко применен и «галоидангидридный» способ активации карбоксильной группы. Были использованы как фторангидриды, так и хлорангидриды этих кислот. Фторангидриды получали реакцией кислоты с избытком цианурфторида в присутствии пиридина при комнатной температуре. Фторангидрид образуется примерно за 1 час и его используют без выделения. Хлорангидриды кислот получали реакцией с избытком тионилхлорида в бензоле при комнатной температуре (примерно 2 ч), избыток тионилхлорида удаляли в вакууме. Синтезированные галоидангидриды кислот конденсировали со спиртом в присутствии ДМАП. Через галоидангидриды получали также и этиленгликолевые эфиры, которые нитрованием азотной кислотой превращали в нитроэтиленгликолевые эфиры.
2.6.3. Химические свойства 1,3-ДНГ-эфиров ПГ.
Химические свойства синтезированных 1,3-ДНГ-эфиров ПГ изучены в реакциях химического перехода между ПГ различных типов и получения их производных, в частности в реакции гидроксиаминометилирования.
Схема 15
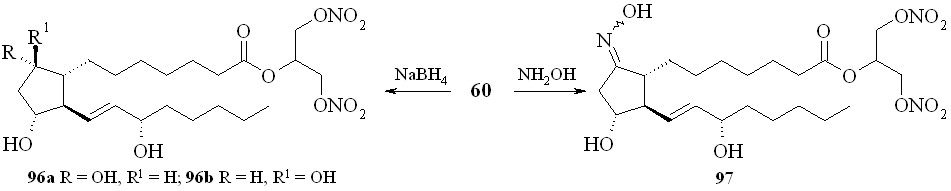
Схема 16
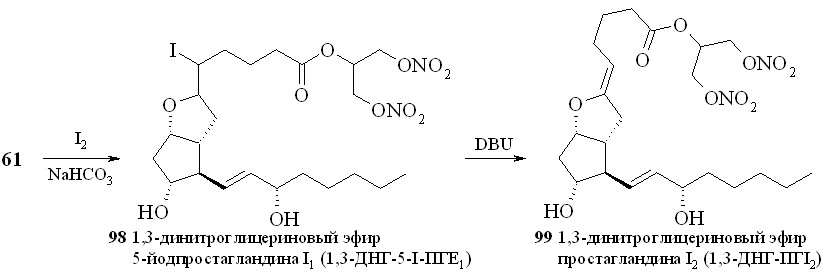
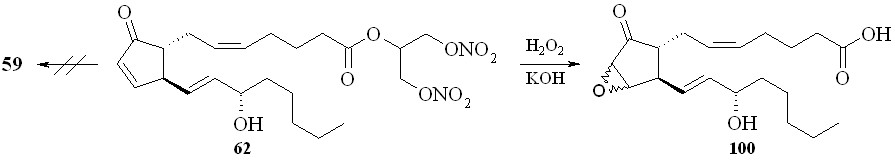
Восстановление кетогруппы простагландинов типа E позволяет перейти к простагландинам типа F. Так, из 1,3-ДНГ-ПГE1 (60) действием боргидрида натрия в метаноле были получены 1,3-ДНГ-ПГF1 (96а,b) в виде смеси - и -изомеров, что свидетельствует об устойчивости нитрогруппы глицериновой части молекулы в реакции восстановления данным реагентом (схема 15). Соотношение получаемых изомеров практически равное, с очень незначительным преобладанием -изомера. 1,3-ДНГ-эфиры других менее доступных простагландинов также могут быть получены по стандартным методам превращений между типами простагландинов (схема 16). Так, 1,3-ДНГ-эфир ПГI2 (1,3-ДНГ-эфир простациклина) (99) синтезирован из эфира (61) циклизацией с йодом в эфир (98) с последующим дегидроиодированием в присутствии 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU). Реакцией с гидроксиламином из эфира (60) получен 1,3-ДНГ-эфир 9-оксиимино-ПГE1 (1,3-ДНГ-ПГE1-ОХ) (97), также в виде смеси син- и анти-изоме-ров (схема 11). Однако в отличие от восстановления кето-группы, здесь реакция проходит с преобладанием анти-изомера. Следует отметить, что попытки получить 1,3-ДНГ-эфиры 9-оксииминопростагландинов E1 и E2 реакцией самих 9-оксииминопростаглан-динов с 1,3-динитратом глицерина (5) давали гораздо худший результат: выход конечного продукта драматически падал независимо от применяемого способа этерификации. В щелочных условиях 1,3-ДНГ-эфиры ПГ не устойчивы. Так, нам не удалось получить 1,3-ДНГ-эфир ПГE2 (59) из 1,3-ДНГ-эфира ПГА2 (62). По стандартной процедуре превращения простагландинов типа A в тип E первая стадия заключается в эпоксидировании двойной связи в циклопентановом кольце молекулы ПГ действием перекиси водорода в присутствии гидроокиси калия. Оказалось, что в этих условиях из 1,3-ДНГ-эфира ПГА2 (62) получается исключительно 10,11-эпокси-ПГА2 (100) (схема 17), а не его ДНГ-эфир. Специальным экспериментом (инкубирование 1,3-ДНГ-эфира ПГА2 (62) с H2O2 в отсутствии КОН) показано, что сама перекись водорода без основного катализа не вызывает деградации ДНГ-эфира или его гидролиза.
Схема 17
3. Биологические свойства синтезированных соединений.
3.1. Биологические свойства 1,3-динитроглицериновых и нитроэтиленгликолевых эфиров ПНЖК.
Была изучена антиагрегационная активность синтезированных 1,3-ДНГ-эфиров ПНЖК 81, 82, 84–87 (табл. 2). Наиболее выраженную антиагрегационную активность в случае АК-индуцированной агрегации проявил 1,3-ДНГ-ДГК (82). В случае АДФ-индуцированной агрегации этот эфир также заметно снижал способность тромбоцитов к взаимодействию друг с другом. 1,3-ДНГ-АК (81) в отличие от самой свободной АК, которая является проагрегантом, не индуцировал агрегацию тромбоцитов. Он эффективно ингибировал как АК-, так и АДФ-индуцированную агрегацию тромбоцитов при концентрации 0,1 мг/мл. Таким образом, включение в молекулу АК динитроглицеринового фрагмента привело к полной потере этой кислотой проагрегационных свойств. 1,3-ДНГ-эфиры других жирных кислот также в той или иной степени ингибировали межтромбоцитарное взаимодействие (табл. 2).
Табл. 2. Влияние 1,3-ДНГ-эфиров жирных кислот на агрегацию тромбоцитов
человека in vitro, индуцированную АК (1) и АДФ (2) (Аmax, %)
| Соединение | Концентрация исследуемого вещества, мг/мл | |||||||
| 0 (контроль) | 0.1 | 0.01 | 0.001 | |||||
| 1 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | |
| 1,3-ДНГ-АК (81) | 75±3 | 79±3 | 12±3 | 26±2 | 50±3 | 58±3 | ||
| 1,3-ДНГ-ДГК (82) | 54±3 | 64±4 | 16±2 | 48±5 | 23±1 | 67±7 | 43±3 | |
| 1,3-ДНГ-ДМАК (84) | 64±3 | 69±4 | 48±3 | 59±2 | 63±1 | 65±5 | ||
| 1,3-ДНГ-ЛНК (85) | 55±2 | 61±8 | 47±3 | 47±3 | 57±3 | 57±3 | ||
| 1,3-ДНГ-ЛНЛК (86) | 54±3 | 59±6 | 30±3 | 39±6 | 42±3 | 42±3 | 46±2 | 46±2 |
| 1,3-ДНГ-ПК (87) | 53±3 | 59±1 | 29±4 | 56±2 | 51±2 | 57±2 | 48±5 | 57±2 |
Было показано, что НЭГ-АК (92) обладает выраженной каннабимиметической активностью. Во всех четырех тестах классической «каннабиноидной тетрады» НЭГ-эфир (92) проявил каннабиноидоподобное действие, сходное с анандамидом. Он дозозависимо вызывал аналгезию (тест «горячая пластинка»), каталепсию (тест с кольцом), гипотермию и резко снижал локомоторную активность (тест «открытое поле») (табл. 3). (Эксперименты проведены под руководством к.х.н. М.Ю.Боброва).
Табл. 3
| Тест «каннабиноидной тетрады» | Контроль | НЭГ-АК |
| «Горячая пластинка» (время задержки болевой реакции), % | 100 | 159 ± 26 |
| «Открытое поле» (число секторов, пересе-ченных с 3-й по 15-ю мин после инъекции) | 57.6 ±11.5 | 4.6 ± 1.5 |
| «Кольцо» (время в неподвижности в течение 5 мин наблюдения), с | 29.5 ±7.5 | 185.5 ± 15.4 |
| Падение ректальной температуры через 10 мин после инъекции, °С | – | –2.62 ± 0.5 |
Изучено взаимодействие 1,3-ДНГ-АК (81) и НЭГ-АК (92) с оксигемоглобином (HbO2) и метгемоглобином (mHb). (Эксперименты проведены под руководством д.б.н. М.А.Киселя). Показано, что добавление эфира (81) к HbO2 сопровождается существенным увеличением скорости окисления гемопротеина в высокоспиновую ферриформу – mHb. Уже при соотношении гем : эфир = 1:2 значительная доля HbO2 превращается в mHb. Мононитрат НЭГ-АК (92) не оказывал подобного действия. Динитрат 1,3-ДНГ-АК (81) вызывал также изменение и спектральных характеристик метгемоглобина, что, возможно, связано с нарушением целостности молекулы белка и потерей гема.
Изучено влияние этиленгликолевого (ЭГ-ДГК) и нитроэтиленгликолевого (НЭГ-ДГК) (93) эфиров докозагексаеновой кислоты на калиевые потенциалозависимые каналы и АМРА-рецепторы – одного из трех подтипов глутаматэргических рецепторов – и на функционирование изолированных митохондрий печени крыс. Оба эфира не влияли на потенциалозависимые калиевые каналы в отличие от действия самой докозагексаеновой кислоты. В то же время они оказывали заметное влияние на ответы АМРА рецепторов, хотя характер этого влияния был отличен для каждого вещества. Если ЭГ-ДГК вызывал дозозависимую потенциацию трансмембранных КК-вызванных токов (КК – каиновая кислота – агонист АМРА рецепторов) в нейронах Пуркинье мозжечка крыс, то его нитроксианалог (93) вызывал, наоборот, их дозозависимое угнетение (табл. 4). (Эксперименты проведены под руководством д.б.н. В.В.Григорьева). Таким образом, введение NO-донорного фрагмента изменило характер действия ЭГ-ДГК на противоположный.
Таблица 4. Действие производных докозагексаеновой кислоты на амплитуды
каинат-вызванных токов в нейронах Пуркинье мозжечка крыс
| Концентрация | % изменения токов АМРА рецепторов* | |
| ЭГ-ДГК | НЭГ-ДГК (66) | |
| 10 нМ | +15% | –11% |
| 100 нМ | +79% | –24% |
| 1 мкМ | +62% | –24% |
* амплитуда каинат-вызванных токов в отсутствие производных докозагексаеновой кислоты взята за 100%.
ЭГ-ДГК и НЭГ-ДГК (93) при добавлении к суспензии митохондрий, не содержащей кальция, вызывали их деполяризацию. Они также дозозависимо предотвращали кальций-индуцированное набухание митохондрий. В присутствии циклоспорина А – ингибитора неспецифической проницаемости мембран – этот эффект усиливался. (Эксперименты проведены под руководством к.х.н. Е.Ф.Шевцовой).
3.2. Биологические свойства 1,3-динитроглицериновых эфиров ПГ.
Исследование биологических свойств синтезированных 1,3-динитроглицериновых эфиров простагландинов показало, что введение в молекулу простагландина NO-донорного фрагмента – 1,3-динитроглицериновой группировки резко изменило фармакологический профиль последних. Так, 1,3-ДНГ-ПГE2 (59) в 5 раз более активен как гипотензивный агент, чем сам ПГЕ2 (табл. 5). При этом он не вызывает изменения частоты сердечных сокращений и тахифилаксии у подопытных животных. Еще одним важным отличием эфира (59) является его способность расслаблять гладкие мышцы изолированной аорты крысы, тогда как ПГE2 является вазоконстриктором. Причем этот эффект не зависит от типа агониста, которым было вызвано предварительное сокращение изолированной аорты (адреналин, ПГE2 или ПГF2) (табл. 6). Для 1,3-ДНГ-ПГE2 (59) также характерно значительное увеличение (более чем в 20 раз) бронходилататорной активности и уменьшение констрикторной активности по отношению к изолированной матке по сравнению с ПГE2 (табл. 5). (Эксперименты по изучению вазодилататорной и миорелаксантной активностям синтезированных соединений проведены под руководством к.м.н. В.В.Малыгина).
Таблица 5. Биологические свойства 1,3-ДНГ-ПГE2
| Фармакологический тест | Активность, EC50, M-6 | |
| 1,3-ДНГ-ПГE2 | ПГE2 | |
| Снижение кровяного давления (ED20) | 2.7±0.13 | 13.1±0.2 |
| Сокращение изолированной матки крысы | 0.4±0.1 | 0.08±0.016 |
| Изолированная аорта крысы | 0.68±0.12 (расслабление) | 84±1 (сокращение) |
| Расслабление трахеи морской свинки | 0.007±0.025 | 0.14±0.08 |
| Сокращение дна желудка крысы | 0.06±0.01 | 0.04±0.015 |
Таблица 6. Вазодилататорная активность 1,3-ДНГ-ПГE2.
| Тип агониста | Расслабление изолированной аорты крысы, EC50, M-6 | |
| 1,3-ДНГ-ПГE2 | 1,3-ДНГ + ПГE2 | |
| адреналин | 1.7±0.15 | 60.0±40.0 |
| ПГЕ2 | 0.68±0.31 | 3.6±1.0 |
| ПГF2 | 3.0±1.0 | 8.1±1.0 |
Аналогично изменились и фармакологические свойства ПГF2. 1,3-ДНГ-ПГF2 (61) более чем в 10 раз превосходит ПГF2 по способности сокращать миометрий матки крысы. При этом он менее активен, чем исходный ПГF2, как констриктор гладких мышц желудка крысы и не отличается от него по действию на гладкие мышцы кишечника. Эфир (61) также является вазодилататором по отношению к изолированным аорте крысы и трахее морc-кой свинки, тогда как сам ПГF2 обладает вазоконстрикторными свойствами по отношению к этим объектам (табл. 7).
Введение NO-донорного фрагмента резко изменило фармакологические свойства и ПГЕ1. 1,3-ДНГ-ПГE1 (60) и 1,3-ДНГ-ПГE1-ОХ (97), так же как и предыдущие 1,3-ДНГ-эфиры ПГ (59 и 61), являются вазодилататорами, а сам ПГЕ1 – вазоконстриктор. При этом следует отметить, что и 9-оксииминопростагландин E1 (ПГE1-ОХ) оказался вазодилататором, хотя и намного более слабым (примерно два порядка), чем динитроглицериновые эфиры. Скорее всего, это связано с тем, что и оксииминная группировка является донором окиси азота, но гораздо менее эффективным, чем нитроксигруппа. Миотропная активность по отношению к изолированной матке крысы у эфира (60) выше, а констрикторное действие на желудок почти на порядок слабее, чем у природного ПГE1 (табл. 8). Последнее свойство позволяет преодолеть одно из ограничений природных ПГ как лекарственных препаратов, а именно их констрикторное действие на желудочно-кишечный тракт, приводящее к диарее.
Таблица 7. Биологические свойства 1,3-ДНГ-ПГF2
| Фармакологический тест | Активность, EC50, M-6 | |
| 1,3-ДНГ-ПГF2 | ПГF2 | |
| Сокращение изолированной матки крысы | 0.009±0.0017 | 0.11±0.04 |
| Изолированная аорта крысы | 0.54±0.19 (расслабление) | (сокращение) |
| Изолированная трахея морской свинки | 10±1.5 (расслабление) | (сокращение) |
| Сокращение дна желудка крысы | 0.13±0.01 | 0.05±0.015 |
| Сокращение изолированной кишки крысы | 0.175±0.07 | 0.13±0.071 |
Таблица 8. Биологические свойства 1,3-ДНГ-ПГE1 и 1,3-ДНГ-ПГE1-ОХ
| Фармакологи-ческий тест | Активность, EC50, M-6 | |||
| 1,3-ДНГ-ПГE1 | ПГE1 | 1,3-ДНГ-ПГE1-ОХ | ПГE1-ОХ | |
| Сокращение матки крысы | 0.33±0.08 | 2.70±0.80 | ||
| Изолированная аорта крысы | 2.10±1.50 (расслабление) | 0.16±0.11 (сокращение) | 0.64±0.17 (расслабление) | 54.16±45.0 (расслабление) |
| Сокращение дна желудка крысы | 0.30±0.012 | 0.04±0.01 | ||
Наблюдаемые изменения фармакологической активности синтезированных 1,3-ДНГ-эфиров ПГ, по-видимому, связаны именно с введением в молекулу NO-донорного фрагмента, а не глицеринового остатка. Известно, что глицериновые эфиры простагландинов являются слабыми агонистами «классических» ПГ-рецепторов, но в то же время обладают собственной фармакологической активностью, практически не блокируемой антагонистами ПГ-рецепторов. Данные по воздействию 1,3-глицериновых эфиров ПГ на гладкие мышцы отсутствуют в литературе, но, учитывая отмеченное выше слабое взаимодействие с ПГ-рецепторами этих эфиров, можно предположить, что значительный вклад в изменение спектра миотропной активности вносит введение в молекулу глицеринового эфира NO-донорного фрагмента. Особенно это заметно по выявленному у 1,3-ДНГ-эфиров ПГ мощному вазодилататорному действию. При этом инкубация изолированной аорты крысы с эквимолярной смесью ПГE2 и 1,3-динитрата глицерина (5) также вызывает релаксацию гладких мышц аорты, а не констрикцию, которую индуцирует сам ПГE2. Следует, однако, заметить, что данная смесь значительно уступает по своей релаксантной активности 1,3-ДНГ-ПГE2 (59) (табл. 6).
ПГЕ1 проявляет сильную антиагрегационную активность. Аналогичными свойствами обладает и окись азота. Поэтому представлялось весьма инте
Табл. 9. Влияние 1,3-ДНГ-ПГE1 и 1,3-ДНГ-ПГE1-ОХ на агрегацию тромбоцитов
человека in vitro, индуцированную АДФ (10-5 М) (Аmax, %)
| Соединение | Концентрация исследуемого вещества, мг/мл | |||||||
| 0 контроль | 10 | 1 | 0.1 | 0.01 | 0.001 | 110-4 | 110-5 | |
| PGE1 | 60±3 | 12±1 | 15±2 | 27±4 | 43±5 | 50±1 | 54±4 | |
| ПГЕ1-ОХ | 76±1 | 7±1 | 58±4 | 63±3 | 73±3 | 69±2 | 67±2 | |
| ДНГ-ПГЕ1 | 56±2 | 7±1 | 12±1 | 29±4 | 37±4 | 47±1 | 51±3 | |
| ДНГ-ПГЕ1-ОХ | 76±1 | 4±1 | 19±2 | 64±4 | 67±5 | |||
ресным выяснить, какое влияние окажет введение NO-донорно гофрагмента именно на это свойство ПГЕ1. Проведенные эксперименты показали, что 1,3-ДНГ-ПГE1 (60) и 1,3-ДНГ-ПГE1-ОХ (97) обладают выраженными антиагрегационными свойствами. Они дозозависимо ингибирует агрегацию тромбоцитов, вызванную арахидоновой кислотой (АК) и аденозиндифосфатом (АДФ). Наиболее ярко это свойство проявляется у эфира 60. Он не намного, но все-таки лучше ингибирует агрегацию, чем сам ПГЕ1 (табл. 9). (Эксперименты по изучению антиагрегационной активности синтезированных соединений проведены под руководством д.м.н. В.А.Макарова).
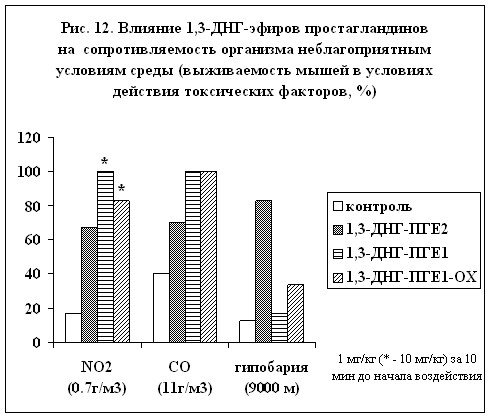
Было изучено влияние 1,3-ДНГ-эфиров ПГ на сопротивляемость организма неблагоприятным условиям среды (рис. 12). Проведенные исследования показали, что 1,3-ДНГ-эфиры ПГ на модели гипобарической гипоксии проявляют защитную активность. Наиболее ярко это свойство проявилось у 1,3-ДНГ-ПГЕ2 (59). При токсической гипоксии (моделировали острым воздействием окиси углерода) наилучшее защитное действие отмечено у 1,3-ДНГ-ПГE1 (60) и 1,3-ДНГ-ПГE1-ОХ (97). Они же оказались наиболее эффективными и при защите от токсического отека легких с развитием дыхательной недостаточности (моделировали острым воздействием диоксида азота), хотя эти соединения применялись в концентрации в 10 раз большей, чем 1,3-ДНГ-ПГЕ2 (59). (Эксперименты проведены под руководством д.м.н. В.В.Чумакова).
На основе 1,3-ДНГ-ПГE1 (60) нами были разработаны мицеллярные и липосомальные композиции, улучшающие локальное кровообращение. Полученные композиции позволяют создать высокую локальную концентрацию ДНГ-эфира в месте нанесения препарата, необходимую для достижения терапевтического эффекта, путем ограничения его распространения с кровотоком, и тем самым предохраняя его от быстрой биодеградации в организме. Этот эффект особенно проявляется при использовании липосомальной формы, то есть когда 1,3-ДНГ-ПГE1 (60) при создании лекарственной формы предварительно включается в липосомы из природного фосфатидилхолина. Специальными экспериментами
на животных (кролики) было показано локальное действие препарата. Так, было отмечено отсутствие снижения агрегационной способности тромбоцитов в общем кровотоке, а также минимальное воздействие на систему гемодинамики и гемостаз. При этом разработанные композиции показали очень хороший результат при лечении
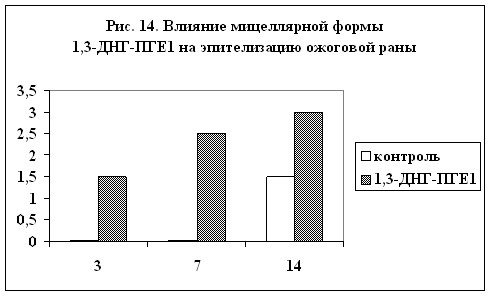
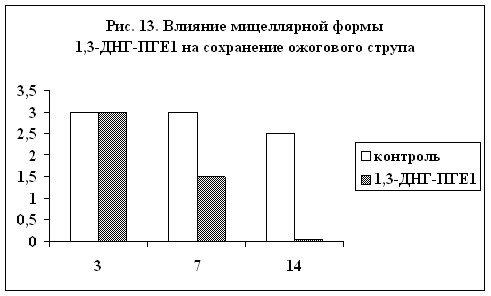
ожоговых поражений. На рис. 13 и 14 показано влияние мицеллярной формы 1,3-ДНГ-ПГE1 (60) на сохранение ожогового струпа и на эпителизацию ожоговой раны (результаты выражены в условных единицах, максимальная выраженность признака – 3). Из диаграмм видно, что при применении препарата, содержащего 1,3-ДНГ-ПГE1 (60), заживление ожоговой раны происходит гораздо быстрее. Через две недели струп полностью отпадает, а под ним обнаруживается молодая эпителиальная ткань без признаков кератизации. В контроле же струп практически сохранялся, а по его краям появлялись следы нагноения.
7. Заключение.
Нами разработаны основные подходы к созданию гибридных физиоло-гически активных соединений, содержащих NO-донорный фрагмент, на основе нитратов биологически активных спиртов как основы потенциальных полифункциональных высокоэффективных лекарственных препаратов. Раз–работаны способы введения NO-донорной группы в молекулу ПГ и ПНЖК как с помощью линкера, так и в безлинкерном варианте. В качестве линкеров нами предложены биологически активные спирты, такие как глицерин, эти–ленгликоль, ряд аминоспиртов. Они после превращения в нитроксисоедине–ния образовывали NO-донорный фармакофор, который присоединяли к мо–лекуле природных веществ (ПГ или ПНЖК). Разработанные способы введе–ния NO-донорного фрагмента, а также сами NO-донорные группировки на основе биологически активных спиртов оказались весьма удобными и уни–версальными. С их использованием синтезированы NO-линкерные гибрид–ные соединения на основе антибиотиков и NSAID. Разработаны способы синтеза гибридных соединений, у которых NO-генерирующая группа прис–оединялась к молекуле исходного вещества непосредственно. Такие безлин–керные соединения синтезированы на основе простагландинов, полиненасы–щенных жирных кислот, гидроксиаминокислот и ряда других биологически активных соединений.
Таким образом, на основании концепции гибридных NO-содержащих соединений разработаны универсальные способы синтеза таких гибридных соединений и синтезирована обширная библиотека нитратов биологически важных природных веществ и действующих начал известных синтетических лекарственных веществ. Эти исследования, инициированные предложенным нами синтезом динитроглицериновых эфиров простагландинов, в настоящее время превратились в одно из активно развиваемых направлений дизайна потенциальных полифункциональных лекарственных препаратов.
Биологические испытания подтвердили положительные изменения фармакологических свойств полученных гибридных соединений по сравне–нию с исходными веществами. Так, введение NO-донорного фрагмента усилило специфическую вазодилататорную, бронхолитическую и миотропную (матка) активности природных ПГ и одновременно снизило их констрикторное действие на желудочно-кишечный тракт. Это делает данные соединения перспективными для создания на их основе бронхолитических препаратов и препаратов для родовспоможения. Введение динитроглицеринового фрагмента в молекулу АК превращает ее из проагреганта в антиагрегант. Синтезированные 1,3-ДНГ-эфиры ПГ показали хорошие результаты по защите экспериментальных животных от воздействия вредных химических факторов. На основе 1,3-ДНГ-ПГЕ2 разработаны композиции, показавшие высокую противоожоговую активность.
Таким образом, синтезированные гибридные соединения, содержащие NO-донорный фрагмент, являются весьма перспективными в плане создания на их основе лекарственных многофункциональных препаратов. Эти соединения могут найти применение в качестве бронхолитических препаратов, в акушерстве, лечебной косметологии, как противоожоговые средства экстренной терапии, особенно когда ожоговое поражение сопровождается отравлением продуктами горения. Отдельные положения диссертации защищены отечественными и зарубежными патентами.
ВыВОДы
- На основе созданной концепции гибридных соединений, содержащих NO-донорный фрагмент, разработаны общие схемы их синтеза как с помощью линкеров, несущих NO-донорную группу, так и в безлинкерном варианте.
- Показана универсальность разработанных методов введения NO-донорного фрагмента на основе биологически активных спиртов.
- Разработаны способы синтеза гибридных соединений на основе простагландинов и полиненасыщенных жирных кислот, содержащих нитроксигруппу в качестве NO-донорного фрагмента.
- Впервые предложено использовать триметилсилильную защиту в синтезе фторангидридов простагландинов.
- Впервые описано нитрование аллильной гидроксильной группы в молекуле простагландина и синтезированы 15-нитраты 11-дезокси-ПГЕ1 и его метилового эфира.
- Синтезированы новые гибридные соединения, содержащие нитроксигруппу, на основе антибиотиков и ряда нестероидных противовоспалительных средств.
- Разработаны способы синтеза нитратов гидроксиаминокислот и впервые синтезированы нитраты L- и D-серина и L-треонина, а также дипептиды на их основе.
- Проведенные биологические исследования на моделях in vitro и in vivo показали, что добавление NO-донорных группировок в молекулу ПГ и ПНЖК резко меняет профиль действия последних, усиливая фармакологически полезные свойства и снижая побочные эффекты. Так динитроглицериновый эфир ПГЕ2 показал увеличенную более чем в 20 раз бронхолитическую активность по сравнения с немодифицированным ПГЕ2, а динитроглицериновый эфир ПГF2 на порядок превосходил природный ПГF2 как констриктор мышц изолированной матки крысы. Введение динитроглицериновой группы в молекулу природного простагландина превратило последние из вазоконстрикторов в вазоделататоры по отношению к препаратам изолированной аорты. Введение динитроглицериновой группы в молекулы полиненасыщенных жирных кислот придаёт им способность ингибировать агрегацию тромбоцитов человека. Включение в молекулу арахидоновой кислоты динитроглицеринового фрагмента привело к полной потере этой кислотой проагрегационных свойств и превратило ее в антиагрегант.
- Разработанная на основе 1,3-динитроглицеринового эфира ПГЕ1 композиция показала эффективные результаты при лечении ожоговых травм у экспериментальных животных.
ОСНОВНые РЕЗУЛЬТАТы ДИССЕРТАцИИ
ОПУБЛИКОВАНы В СЛЕДУЮЩИХ РАБОТАХ
Патенты
- Серков И.В., Безуглов В.В., Пачева Л.М., Малыгин В.В., Гафуров Р.Г., Лилле Ю.Э., Самель Н.Э., Бергельсон Л.Д. 1',3'-Динитроглицериновый эфир проcтагландина F2, обладающий миотропной активностью по отношению к гладкой мускулатуре // Авторское свидетельство № 1640963, приоритет от 06.10.1989.
- Безуглов В.В., Серков И.В., Пачева Л.М., Голованова Н.К., Журавлева Л.И., Самель Н.Э., Лилле Ю.Э., Малыгин В.В., Безноско Б.К., Гафуров Р.Г., Бергельсон Л.Д. 1',3'-Динитроглицериновый эфир проcтагландина Е2, обладающий гипотензивной вазо- и бронходилаторной активностью // Авторское свидетельство № 1832680, приоритет от 06.10.1989.
- Серков И.В., Безуглов В.В., Пачева Л.М., Петрухина Г.Н., Самель Н.Э., Макаров В.А., Малыгин В.В., Лилле Ю.Э., Гафуров Р.Г., Бергельсон Л.Д. 1',3'-Динитроглицериновый эфир проcтагландина Е1 и 9-оксима проcтагландина Е1, обладающие вазодилаторной и антиагрегационной активностями // Авторское свидетельство № 1825786, приоритет от 15.01.1991.
- Безуглов В.В., Серков И.В. 1,3-Динитроглицериновые эфиры полиненасыщенных жирных кислот, гидроксипроизводных полиненасыщенных жирных кислот и простагландинов и способы их получения // Патент РФ № 2067094, приоритет от 27.09.1993 (Бюл. № 27, 27.09.96).
- Безуглов В.В., Серков И.В., Дмитриев П.И., Воложин А.И., Петрухина Г.Н., Макаров В.А. Средство, улучшающее кровообращение, для наружного применения // Патент РФ № 2098097, приоритет от 07.07.1994.
- Bezuglov V.V., Serkov I.V. Dinitroglycerol esters of prostaglandins // US Patent № 5,625,083, 29.04.1997.
- Серков И.В., Безуглов В.В. Нитроксиалкиламинокислоты // Патент № 2340597, приоритет от 05.06.2008.
Статьи
- Безуглов В.В., Бобров М.Ю., Грецкая Н.М., Арчаков А.В., Серков И.В., Феденюк А.П., Веревочкина Е.Ю., Когтева Г.С., Титова О.Ю., Марва-нов Д.М., Де Петроцельс Л., Бизоньо Т., Ди Марцо В., Маневич Е.М. Арахи-доноилэтиленгликоль и его нитроэфир – новые каннабимиметические соеди-нения: окисление 15-липоксигеназой и гидролиз гидролазой амидов жирных кислот // Биоорганическая химия. – 1998. – Т. 24. – N 12. – С. 953957.
- Безуглов В.В., Андреюк Г.М., Серков И.В., Кисель М.А. Влияние липид-ных производных динитроглицерина и нитроэтиленгликоля на спектральные параметры гемоглобина человека // Биохимия. – 2000. – Т. – 65. – Вып. 6. – С. 804809.
- Васильева Т.М., Петрухина Г.Н., Макаров В.А., Серков И.В., Грецкая Н.М., Безуглов В.В. Действие новых синтетических динитроглицериновых эфиров жирных кислот на агрегацию тромбоцитов человека // Эксп. Клин. Фармакология. – 2003. – Т. 66. – № 6. – С. 44–46.
- Серков И.В., Безуглов В.В. O-Нитрование в простагландинах: синтез
15-O-нитрата-11-дезоксипростагландина E1 и его метилового эфира // Биоор-ганическая химия. – 2006. – Т. 32. – № 1. – С. 110112.
- Серков И.В., Григорьев В.В., Иванова Т.А., Грецкая Н.М., Безуглов В.В., Бачурин С.О. Действие производных докозагексаеновой кислоты на АМРА рецепторы в нейронах Пуркинье // Доклады Академии наук. – 2006. – Т. 411. – № 3. – С. 1–2.
- Серков И.В., Безуглов В.В. Синтез новых эфиров и амидов цефалоспо-рина G // Химия природных соединений. – 2007. – № 1. – С. 85–88.
- Серков И.В., Шевцова Е.Ф., Дубова Л.Г., Киреева Е.Г., Вишневская Е.М., Грецкая Н.М., Безуглов В.В., Бачурин С.О. Взаимодействие производных докозагексаеновой кислоты с митохондриями // Доклады Академии наук. –2007. – Т. 414. – № 3. – С. 14.
- Серков И.В., Безуглов В.В. О-Нитраты гидроксиаминокислот серина и треонина // Химия природных соединений. – 2008. – № 1. – С. 52–53.
- Серков И.В., Безуглов В.В. Фторангидриды простагландинов в синтезе производных природных простагландинов по карбоксильной группе // Биоорганическая химия. – 2009. – Т. 35. – № 1. – С. 1–7.
- Серков И.В., Безуглов В.В. 1,3-O-нитраты циклооксигеназных метаболи-тов эндоканнабиноида 2-арахидоноилглицерина. Синтез и свойства // Биоор-ганическая химия. – 2009. – Т. 35. – № 2. – С. 245–252.
- Серков И.В., Безуглов В.В. Нитроксиалкиламиды как прототипы гибрид-ных нестероидных противовоспалительных препаратов, содержащих NO-донорный фрагмент // Доклады Академии наук. – 2009. – Т. 425. – № 6. – С. 777–779.
- Серков И.В., Безуглов В.В. Многофункциональные соединения, содержа-щие органические нитраты, – прототипы гибридных лекарственных препара-тов // Успехи химии. – 2009. – Т. 78. – № 5. – С. 442–465.
- Андрианова Е.Л., Бобров М.Ю., Грецкая Н.М., Зинченко Г.Н., Серков И.В., Фомина-Агеева Е.В., Безуглов В.В. Действие нейролипинов и их синтетичес-ких аналогов на нормальные и трансформированные глиальные клетки // Нейрохимия – 2010. – Т. 27. – № 1. – С. 53–62.
- Григорьев В.В., Серков И.В., Безноско Б.К., Иванова Т.А., Грецкая Н.М., Безуглов В.В., Бачурин С.О. Действие производных арахидоновой и докоза-гексаеновой кислот на АМРА-рецепторы в нейронах Пуркинье и на когни-тивные функции у мышей // Известия РАН. Серия биологическая. – 2010. – № 3. – С. 370–374.
- Серков И.В., Грецкая Н.М., Безуглов В.В. Нитроанандамид, нитропроста–миды Е2 и F2 и их аналоги // Химия природ. соед. – 2010. – № 5. – С. 591–594.
Тезисы
- Makarov V.A., Petrukhina G.N., Volozshin A.I., Serkov I.V., Bezuglov V.V. The influence of NO-PGs on platelet function and microcirculation // 9-th Internatio-nal conference on "Prostaglandins and related compounds". – Florence, Italy. 4–8 June 1994. – Abstract book. – P. 61.
- Serkov I.V., Bezuglov V.V. Synthesis and properties of NO-PGs // 9-th Inter-national conference on "Prostaglandins and related compounds". – Florence, Italy. 4–8 June 1994. – Abstract book. – P. 61.
- Bezuglov V.V., Serkov I.V. Design of binary prostaglandin preparation. Ap-proaches and examples // 9-th International conference on "Prostaglandins and related compounds". – Florence, Italy. 4–8 June 1994. – Abstract book. – P.45.
- Malygin V.V., Serkov I.V., Bezuglov V.V., Makhaeva G. 1,3-Dinitroglycerol esters of prostaglandins as new perspective "binary" drugs for pharmacology and medicine // XIV-th International symposium on medicinal chemistry – Maastricht, Netherlands. 8–12 September 1996. – Abstract book. – P. P-3.11.
- Безуглов В.В., Серков И.В., Макаров В.А., Воложин А.И., Кузьмина С.М., Маневич Е.М. Простанит – новое решение старых проблем // Международная научно-практическая конференция «Биологически активные вещества и новые продукты в косметике». – Москва. 26–28 ноября 1996. – Тезисы докладов. – С. 16–17.
- Bobrov M.Yu., Gretskaya N.M., Fedenyuk A.P., Yudushkin I.A., Serkov I.V., Muller A., Bonne C., Durand T., Bezuglov V.V. Novel bioactive amides and esters of polyunsaturated fatty acids closely related to endocannabinoids // 11-th International conference on advances in prostaglandin and leukotriene research: basic science and new clinical applications. – Florence, Italy. 4–8 June 2000. – Abstract book. – P. 94.
- Кисель М.А., Андреюк Г.М., Серков И.В., Безуглов В.В. Гемоглобин – ключевой белок в системе генерации NO из органических нитратов липидной природы // IV Съезд Белорусского общественного объединения фотобиоло-гов и биофизиков «Молекулярно-клеточные основы функционирования биосистем». – Минск. 28–30 июня 2000. – Тезисы докладов – С. 254.
- Безуглов В.В., Серков И.В. Синтез производных цефалоспорина G // I Международная конференция «Химия и биологическая активность азотистых Гетероциклов и алкалоидов». – Москва. 9–12 октября 2001. – Тезисы докладов. – Т. 2, С. 36.
- Serkov I.V., Bobrov M.Yu., Bezuglov V.V. Nitroesters of bioeffector lipids as novel NO-boosted regulators // International symposium on advances in synthetic, combinatorial and medical chemistry. – Moscow, 58 May 2004. – Abstract book. – P. 167.
- Серков И.В., Безуглов В.В. Циклооксигеназные метаболиты эндоканнабиноидов, содержащие NO-донорный фрагмент // XVIII Менделеевский съезд по общей и прикладной химии. – Москва. 23–28 сентября 2007. – Тезисы докладов. – Т. 4. – С. 481.
- Григорьев В.В., Серков И.В., Иванова Т.А., Безноско Б.К., Грецкая Н.М., Безуглов В.В., Бачурин С.О. Действие производных арахидоновой и докозагексаеновой кислот на АМРА рецепторы в нейронах Пуркинье и на память у мышей // III Съезд фармакологов России «Фармакология – практическому здравоохранению». – Санкт-Петербург. 23–27 сентября 2007. – Тезисы докладов. – C. 1–1667.
- Серков И.В., Безуглов В.В. О-Нитраты биологически активных спиртов // Конференция «Органическая химия для медицны». – Черноголовка, Москов-ская область. 7–11 сентября 2008. – Тезисы докладов. – C. 235.
- Серков И.В., Вишневская Е.М., Безуглов В.В. Синтез нитроксиаминокис-лот и пептидов на их основе // IV Российский симпозиум «Белки и пептиды» – Казань. 23–27 июня 2009. – Тезисы докладов. – С. 156.
- Серков И.В., Вишневская Е.М., Грецкая Н.М., Безуглов В.В. Амиды ней-роактивных липидов и их циклооксигеназных метаболитов с нитратами ами-носпиртов // VII Всероссийская научная конференция «Химия и медицина, ОРХИМЕД–2009». – Уфа. 1–5 июля 2009. – Тезисы докладов. – С. 266.
- Серков И.В., Вишневская Е.М., Андрианова Е.Л., Бобров М.Ю., Грецкая Н.М., Безуглов В.В. Нитронейролипины и нитрооксилипины как прототипы новых многофункциональных соединений // VIII Всероссийская конференция «Химия и медицина». – Уфа. 6–8 апреля 2010. – Тезисы докладов. – С. 125–126.
15.Бобров М.Ю., Андрианова Е.Л., Грецкая Н.М., Серков И.В., Безуглов В.В. Нейролипины, простамиды и их синтетические аналоги как перспективные нейропротекторы // 5 Международная конференция «Биологические основы индивидуальной чувствительности к психотропным средствам». – Москва.
1–4 июня, 2010.– Тезисы докладов. – С. 27.