

Окислительная активация в синтезе и превращениях органических соединений серы
На правах рукописи
ШИНКАРЬ ЕЛЕНА ВЛАДИМИРОВНА
ОКИСЛИТЕЛЬНАЯ АКТИВАЦИЯ В СИНТЕЗЕ И ПРЕВРАЩЕНИЯХ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ СЕРЫ
02.00.03 – Органическая химия
АВТОРЕФЕРАТ
диссертации на соискание ученой степени
доктора химических наук
Астрахань – 2012
Работа выполнена в Астраханском государственном техническом университете на кафедре органической, биологической и физколлоидной химии
Научный консультант: доктор химических наук,
профессор
Берберова Надежда Титовна
Официальные оппоненты: доктор химических наук,
профессор
Рябухин Юрий Иванович
доктор химических наук,
профессор
Белоглазкина Елена Кимовна
доктор химических наук,
профессор
Гончаров Владимир Ильич
Ведущая организация: Институт органической химии
им. Н.Д. Зелинского
Защита состоится « 2 » марта 2012 г. в 14.00 часов на заседании диссертационного совета ДM 307.001.04 в Астраханском государственном техническом университете по адресу:
414025, г. Астрахань, ул. Татищева, 16, 2-ой учебный корпус, ауд. 201.
С диссертацией можно ознакомиться в научной библиотеке Астраханского государственного технического университета.
Автореферат разослан « » апреля 2012 г.
| Ученый секретарь диссертационного совета доктор технических наук, профессор |  | Каратун О.Н. |
Общая характеристика работы
Актуальность работы. Введение в молекулы органических соединений атома серы обуславливает существенное возрастание их биологической активности. В настоящее время серосодержащие органические соединения включают в состав антибактериальных, противотуберкулезных и противоопухолевых препаратов. Они находят широкое применение в качестве присадок к моторным маслам и топливам, стабилизаторов резин, пластмасс и полимеров, составляющих пестицидов и гербицидов, отбеливающих средств, пищевых ароматизаторов и т.д.
Большинство методов получения тиопроизводных органических соединений основано на использовании термодинамически стабильных и часто кинетически инертных соединений – алкантиолов и сероводорода, являющихся дешевыми источниками (алкил-)тиильных радикалов. В связи с этим осуществить реакции одностадийного тиолирования ароматических соединений в присутствии RSH (R = H, Alk) при низких температурах достаточно сложно. Имеются примеры синтеза ароматических тиолов на основе SN2 реакции, однако сведения о прямом введении тиогруппы в ароматическое кольцо малочисленны. Для постадийной SН- (SR-) функционализации олефинов из серосодержащих реагентов генерируют радикальные интермедиаты в условиях радиолиза, фотолиза или термолиза. Рециклизация незамещенных пятичленных О-, N-гетероциклических соединений с образованием тиофена и его производных протекает только при 350-450 °С.
Несмотря на неоспоримые успехи в синтезе органических соединений серы, необходим поиск новых эффективных путей их получения при снижении энергозатрат. Одноэлектронное окисление сероводорода, тиолов и сульфанов до катион-радикалов в неводных растворителях открывает принципиально новое направление их превращений в органические производные серы. Отсутствие информации об исследованиях катион-радикальной активации сероводорода (проведены только квантово-химические расчеты[1]
, подтверждающие существование катион-радикала H2S+) определяет новизну настоящей работы.
Предлагаемый подход к получению органических соединений серы базируется на окислительной активации RSH (R = H, Alk, HSn) в результате электрохимического (прямым способом, в присутствии электромедиаторов или сильных оснований) и/или химического окисления (с применением пространственно-затрудненных о-бензохинонов или металлокомплексных соединений). Активация серосодержащих реагентов способствует генерированию интермедиатов: сероцентрированных катион-радикалов [RSH]+ (R = H, Alk, HSn) и продуктов их фрагментации с отщеплением протона.
Развитие созданного научного направления, основанного на принципиально новом подходе к разработке целенаправленных методов получения органических соединений серы посредством окислительной активации сероводорода, тиолов и сульфанов и проведении систематизации полученных результатов с целью выбора оптимальных условий синтеза, является актуальным и перспективным.
Вовлечение высокотоксичных серосодержащих компонентов природного газа и нефти (сероводорода и тиолов) в органический синтез решает, в частности, экологическую проблему утилизации вредных примесей. Предложенные новые способы десульфуризации углеводородного сырья и удаления сероводорода (сульфанов) из серы базируются на выработанных принципах окислительной активации RSH (R = H, Alk, HSn).
Цель работы: разработка нового подхода к реализации реакций сероводорода, тиолов и сульфанов с органическими субстратами, основанного на использовании окислительной активации; осуществление поиска новых систем одноэлектронного окисления серосодержащих соединений RSH (R = H, Alk, HSn); изучение влияния условий реакции (способа активации, времени, температуры, способности различных по природе субстратов к окислению в разных растворителях) на строение и выход получаемых соединений; создание экологически приемлемых методов синтеза целевых соединений при снижении энергозатрат; разработка эффективных способов однореакторного синтеза серосодержащих производных алифатического, ароматического и гетероциклического рядов.
Для достижения поставленной цели в работе решались следующие задачи:
- поиск эффективных путей повышения реакционной способности сероводорода, тиолов и сульфанов посредством их электрохимического и/или химического окисления; изучение условий генерирования нестабильных катион-радикалов и других активных интермедиатов ((алкил-)тиильных и полисульфидных радикалов, а также H+ и H. радикалов), участвующих в реакциях с органическими соединениями;
2) обоснование выбора одноэлектронного окислителя соединений RSH (R = H, Alk, HSn) из ряда: платиновый анод; пространственно-затрудненные о-бензохиноны; моноядерные комплексы никеля, хрома, палладия и платины с N,N-, N,S-, S,S-донорными хиноидными лигандами; электромедиаторы (замещенные ароматические амины); комбинированные системы, включающие анод и химический агент (органическое основание, о-бензохинон, молекулярная сера);
- осуществление электросинтеза серосодержащих производных олефинов, ароматических и О-, N-гетероциклических соединений, относящегося к перспективным методам «зеленой химии» и позволяющего уменьшить негативное влияние химических реагентов на окружающую среду;
- установление оптимальных условий синтеза органических производных серы, подготовка рекомендаций его проведения, необходимых для варьирования спектра основных продуктов изучаемых реакций;
- разработка способов, направленных на повышение эффективности процессов нефте- и газопереработки: демеркаптанизации углеводородного сырья и нефтяных фракций, а также дегазации серы, которая образуется при окислении сероводорода по методу Клауса.
Научная новизна.
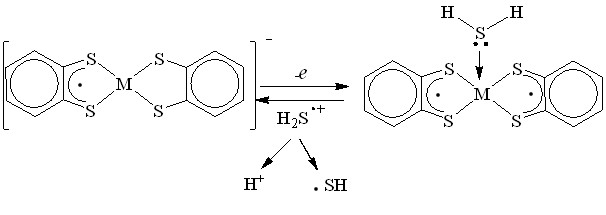
- Впервые продемонстрированы способы генерирования катион-радикалов сероводорода, тиолов и сульфанов прямой электрохимической активацией, действием пространственно-затрудненных о-бензохинонов, комплексов Ni, Сr, Pt, Pd с редокс-активными лигандами, органических электромедиаторов, а также анода и химических агентов (основания Льюиса, молекулярная серы, 3,5-ди-трет-бутил-о-бензохинон). Образование нестабильных катион-радикалов [RSH]+· (R = H, Alk, HSn) доказано методом ЭПР-спектроскопии.
- Проведено комплексное исследование окислительной активации RSH (R = H, Alk, HSn) при их одноэлектронном электроокислении в органических растворителях, определена кислотность катион-радикалов [RSH]+·, подтверждена их способность к фрагментации при комнатной температуре с отрывом протона и образованием (алкил-)тиильных и полисульфидных радикалов в реакциях со слабыми органическими основаниями (пиридином, хинолином, акридином и 2,2’-, 2,3’- и 3,3’-бихинолинами). Изучена SH-функционализация пиридина в присутствии электрохимически окисляемого H2S через стадию протонирования и дальнейшего тиоприсоединения к субстрату.
- Выявлена эффективность активации серосодержащих реагентов в присутствии пространственно-затрудненных о-бензохинонов, комплексов никеля, хрома, палладия и платины с редокс-лигандами (на основе замещенных о-фенилендиамина, о-аминотиофенола, о-фенилендитиола, 4-метил-о-фенилендитиола), способных к регенерации кислородом воздуха; получены алифатические, ароматические и гетероциклические тиолы, сульфиды и дисульфиды на поверхности -Al2O3, SiO2nH2O и Na2OAl2O3xSiO2, модифицированной одноэлектронными окислителями.
- Установлены закономерности окислительной активации сероводорода, тиолов и сульфанов в присутствии электрогенерируемых катион-радикальных форм медиаторов (три-п-толиламин, три-п-бромфениламин, 2,2’,4,4’-тетраметоксидифениламин, N,N,N’,N’-тетраметил-1,4-фенилендиамин, комплексы никеля и хрома с редокс-лигандами) и при использовании комбинированных систем (анод и химический реагент), что позволяет получать органические соединения серы при снижении потенциала электролиза по сравнению с методом прямого электросинтеза.
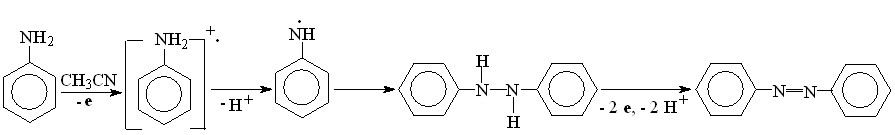
- Впервые получен ряд тиопроизводных ароматических соединений (бензол, толуол, фенол, нитробензол, бензойная кислота) прямым введением тиогруппы в ароматическое ядро; показана принципиальная возможность синтеза ряда тиопиранов и тиопирилиевых солей из 1,5-карбонильных соединений в условиях электрохимической активации и в присутствии комплексов металлов VIII группы с редокс-лигандами.
- Показана принципиальная возможность управления синтезом соединений тиофенового ряда на основе фурана, 2,5-диметилфурана и пиррола с участием окисленной формы сероводорода электрохимическим и/или химическим способом. Впервые проведена замена атомов кислорода и азота в пятичленном цикле на серу при 25 °С и осуществлены превращения тиофена в 2-тиофентиол, бис(2-тиенил)сульфид, бис(2-тиенил)дисульфид, олиго- и политиофены.
Практическая значимость работы. Разработан ряд новых, препаративно удобных и управляемых методов синтеза органических соединений серы на основе электрохимически и/или химически окисленных сероводорода, тиолов и сульфанов в одном реакционном сосуде при 25-100 °С. Предложены перспективные направления практического применения окислительного инициирования органических реакций с участием сероводорода, тиолов и сульфанов. Разработаны эффективные способы демеркаптанизации углеводородного сырья и очистки газовой серы для предприятий нефтегазодобывающей и перерабатывающей промышленности. Методика спектрофотометрического определения сульфанов в товарной газовой сере предложена для качественного контроля продукции. Предложен экологически приемлемый способ утилизации Н2S в серу посредством его окисления на аноде при низких энергозатратах.
На защиту выносятся следующие основные положения:
- редокс-активность сероводорода, тиолов и сульфанов в органических растворителях; электрохимическое (прямое и косвенное) и химическое одноэлектронное окисление сероводорода, алкантиолов и сульфанов;
- электрохимические способы синтеза органических соединений серы с участием окисленных форм сероводорода, алкантиолов и сульфанов;
- препаративные методы синтеза серосодержащих соединений на основе олефинов, ароматических углеводородов, 1,5-дикарбонильных и гетероциклических соединений в присутствии одноэлектронных окислителей сероводорода, алкантиолов и сульфанов, реализуемых в гомогенных условиях;
- синтез органических соединений серы с участием сероводорода, тиолов и сульфанов в присутствии катион-радикалов органических электромедиаторов;
- препаративные методы синтеза продуктов радикального тио-(замещения) присоединения, S-циклизации и S-рециклизации, осуществляемые на алюмооксидных катализаторах, силикагеле или цеолитах, модифицированных хиноидными и металлокомплексными окислителями сероводорода, тиолов и сульфанов;
- пути практического применения предложенных способов активации сероводорода, тиолов и сульфанов для утилизации и удаления серосодержащих компонентов из углеводородного сырья и продукции нефте-, газопереработки.
Личный вклад автора включает выбор темы и постановку проблемы исследований, теоретическое обоснование поставленных задач и методологии их решения, разработку направлений эксперимента, обработку, анализ и интерпретацию полученных результатов при непосредственном участии на всех этапах исследований.
Апробация работы. Основные результаты работы опубликованы в ведущих научных журналах и прошли апробацию на Всероссийских и международных конференциях, симпузиумах и совещаниях: «Металлоорганические соединения – материалы будущего тысячелетия» (III Разуваевские чтения, Нижний Новгород, 2000), «Механизмы реакции и органические интермедиаты» (Санкт-Петербург, 2001), Чугаевская по координационной химии (Ростов-на-Дону, 2001; Кишинёв, 2005 Санкт-Петербург, 2009), «New Approaches in Coordination and Organometallic Chemistry. Look from 21-th Century» (Нижний Новгород, 2002), «Химия органических соединений кремния и серы» (Иркутск, 2001), «Новые химические технологии: производство и применение» (г. Пенза, 2002), Менделеевский съезд по общей и прикладной химии (Казань, 2003; Москва, 2007; Волгоград, 2011); по электрохимии органических соединений «ЭХОС» (Астрахань, 2002; Новочеркасск, 2006; Тамбов, 2010); «Полиядерные системы и активация малых молекул» (Иваново, 2004), международной конференции «From molecules towards materials» (IV Разуваевские чтения, Нижний Новгород, 2005), по химии гетероциклических соединений, посвященная 90-летию со дня рождения проф. А.Н. Коста (Москва, 2005); «Органическая химия от Бутлерова и Бейльштейна до современности», посвященная 145-летию теории строения органических соединений А.М. Бутлерова и 100-летию памяти Ф.Ф. Бейльштейна (Санкт-Петербург, 2006), по органической химии серы «ISOCS-23» (Москва, 2008); «Электрохимия и экология» (Новочеркасск, 2008); «Новые направления в химии гетероциклических соединений» (Кисловодск, 2009; Железноводск, 2011).
Публикации результатов. По теме диссертации опубликовано 102 работы, из них 1 монография, 23 статьи в изданиях, рекомендованных ВАК, получено 5 патентов РФ на изобретения.
Структура и объём работы. Диссертация изложена на ____ страницах машинописного текста, включая введение, литературный обзор, обсуждение результатов, экспериментальную часть, выводы, список использованных источников из _____ наименований, ____ таблиц, _____ рисунка.
Основное содержание работы
Несмотря на то, что полученные в работе органические соединения серы известны, и имеется достаточно много способов их синтеза, примеры органических реакций сероводорода, тиолов и сульфанов при 25 - 100 °С практически отсутствуют. В связи с этим разработаны новые подходы к проведению реакций RSH (R=H, Alk, HSn) с олефинами, ароматическими, 1,5-дикарбонильными и O-, N-содержащих пяти- и шестичленными гетероциклическими соединениями, базирующиеся на окислительной активации реагентов до катион-радикалов.
В качестве ключевых реагентов рассмотрены сероводород, алифатические тиолы ((этан-, изопропан-, бутан-)гексантиол-1) и сульфаны, экстрагированные из газовой серы, полученной по методу Клауса.
В роли окислителей сероводорода, тиолов и сульфанов предложено использовать:
1) платиновый анод;
2) пространственно-затрудненные о-бензохиноны;
3) комплексы металлов VIII группы с редокс-активными лигандами;
4) органические электромедиаторы;
5) оксид алюминия, силикагель или цеолит, модифицированные о-бензохинонами, а также металлокоплексами;
6) комбинированные системы: анод-о-бензохинон, анод-основание Льюиса, о-бензохинон-молекулярная сера.
Ниже представлены новые подходы к синтезу органических соединений серы на основе активированных сероводорода, алкантиолов и сульфанов.
- Электрохимическая активация сероводорода, алкантиолов и сульфанов
в органических растворителях
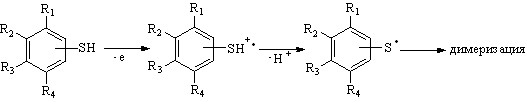
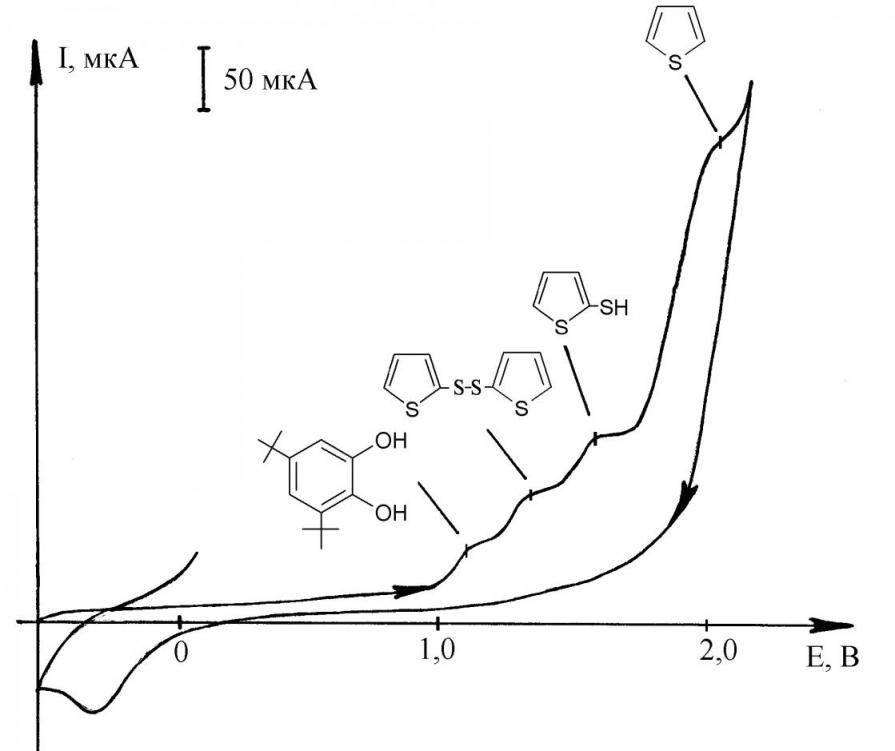
Сероводород, тиолы и сульфаны RSH (R = H, Alk, HSn) в органических растворителях (CH3CN, ДМФА, СН2Сl2) окисляются на платиновом (стеклографитовом) электроде в одну необратимую стадию (рис.1, табл. 1):
![]() (1)
(1)
R = H, С2H5, i-C3H7, n-C4H9, n-C6H13, HSn (n = 2-8)
По методу Бордвелла рассчитаны значения рК нестабильных катион-радикалов RSH+:
![]() (2)
(2)
Повышение кислотности катион-радикалов по сравнению с молекулярной формой соединений RSH (R = H, Alk, HSn) равно рК =30, 16-18, 11, соответственно. При депротонировании катион-радикалов RSH+ образуются (алкил-)тиильные и полисульфидные радикалы, которые димеризуются на поверхности анода.
Табл. 1 Потенциалы окисления и ионизации соединений RSH (R=H, Alk, HSn) (Ag/AgCl, 0,1 М NBu4ClO4 )
Примечание: Е, Е, Е – потенциал окисления в СН3СN на платиновом, в СН3СN на стеклографитовом аноде, в СН2Сl2 на платиновом аноде; ПИ* – потенциал ионизации, рассчитанный по методу РМ3 |
Рис.1 – ЦВА окисления H2S: а) до электролиза; б) после электролиза; идентификация - - - - протона добавкой HClO4, -.-.-.- сульфанов добавкой независимо синтезированных H2Sn (CH3CN, Pt-анод, Ag/AgCl, 0,1 М NBu4ClO4) |
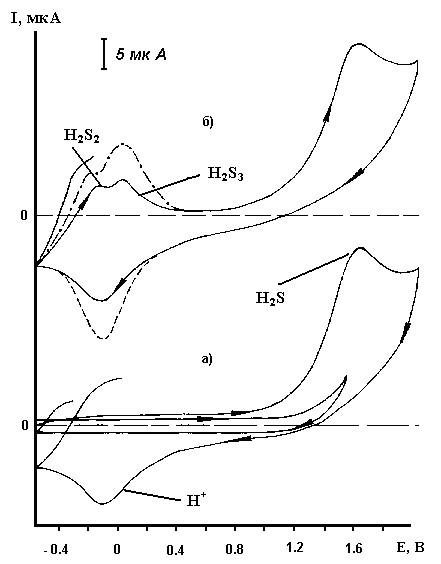

Во фреоновой матрице (77 К) методом ЭПР (рис. 2) зафиксирован триплет (а2Н=29 Гс), отвечающий катион-радикалу Н2S+, время жизни – 15 мин. Значение ПИ(H2S) =10,5 эВ ниже, чем у фреона-113 (11,99 эВ), поэтому происходит отрыв электрона от молекулы H2S. Расщепление спектра
катион-радикала H2S+ связано с димеризацией тиильных радикалов и образованием дисульфанов.
Электролиз соединений RSH (R = H, Alk, HSn) в потенциостатическом режиме при потенциале их окисления приводит к низшим сульфанам (0,2-0,5 В; выход по току 25%), дисульфидам (1,32-1,46 В; выход по току 46%) и молекулярной сере (-0,77 В, выход по току 34%), соответственно.
| Конечным продуктом окисления H2S является сера (выход 63%), идентифицированная методом РФА. При электролизе RSH (R = H, Alk, HSn) на катоде восстанавливается H+ с образованием водорода. Неактивированые на аноде соединения RSH (R = H, Alk, HSn) взаимодействуют со слабыми основаниями Ia-VIIIa (рис. 3) с образованием неустойчивых ат-комплексов (1,43 – 1,56 В), которые разрушаются при 40°C. | 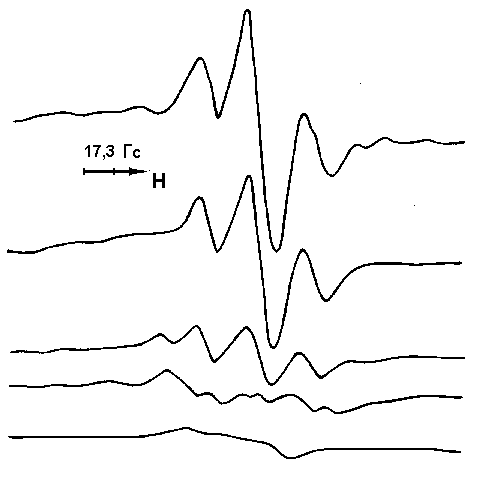 Рис.2 – ЭПР-спектр катион-радикала сероводорода (фреон-113 (CF2ClCFCl2), 0,5% H2S, 77 К) Рис.2 – ЭПР-спектр катион-радикала сероводорода (фреон-113 (CF2ClCFCl2), 0,5% H2S, 77 К) |
 IIa IIa |  IIIa IIIa |  VIa - VIIIa VIa - VIIIa | |
 IVa IVa |  Va Va |
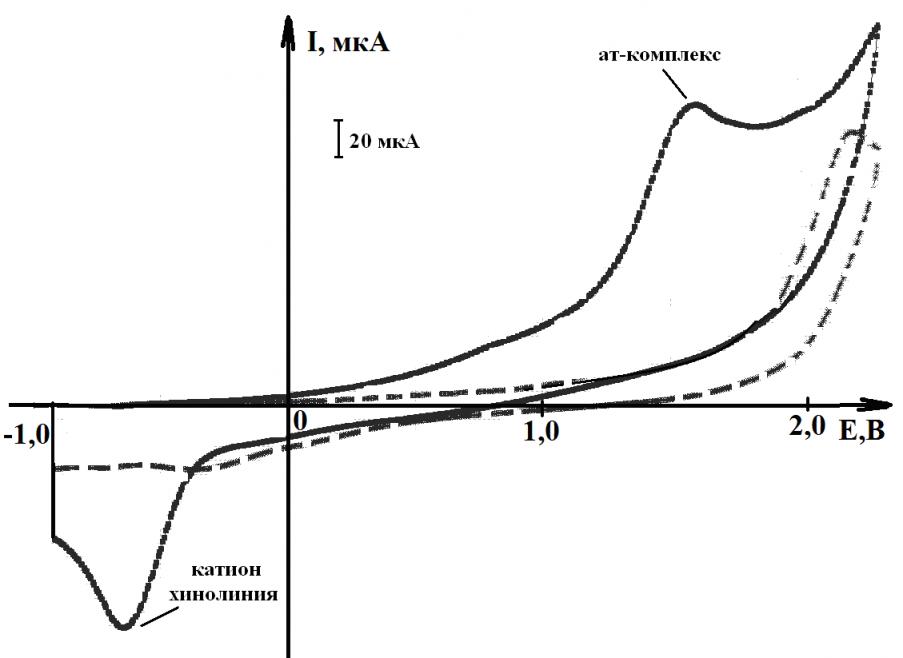 Рис. 3 – ЦВА соединения Ia:_ _ _ а); после добавки Н2S ___ б) (CH3CN, Pt-анод, Ag/AgCl, 0,1 M NBu4ClO4, C(Н2S) = С(Ia) = 510-3 моль/л) Рис. 3 – ЦВА соединения Ia:_ _ _ а); после добавки Н2S ___ б) (CH3CN, Pt-анод, Ag/AgCl, 0,1 M NBu4ClO4, C(Н2S) = С(Ia) = 510-3 моль/л) | В СH3CN, в отличие от ДМФА, электрохимическая активация соединений RSH (R = H, Alk, HSn) в реакциях с основаниями Iа-VIIIа (Епа > 1,7 В) приводит к протонированию субстратов с образованием катионов (Епк = -0,5 – (-0,7) В), что подтверждает высокую кислотность RSH+: 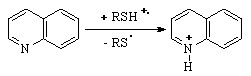 R = H, С2H5, i-C3H7, n-C4H9, n-C6H13, HSn (n = 2-8) (3) R = H, С2H5, i-C3H7, n-C4H9, n-C6H13, HSn (n = 2-8) (3) |
При анодной активации реагентов RSH (R = H, Alk, HSn), также как и в присутствии HClO4, 2,2- и 3,3-бихинолины подвергаются двойному протонированию:
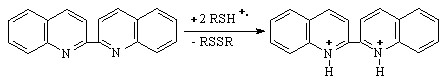 (4)
(4)
При взаимодействии катион-радикалов RSH+ (R = H, Alk, HSn) с основаниями VIа-VIIIа протекает восстановление субстратов до 1’,4’-дигидропроизводных – что на ЭСП фиксируется батохромный сдвиг максимума поглощения (мах=333385 нм). Таким образом, электрохимическое одноэлектронное окисление соединений RSH (R=H, Alk, HSn) приводит к генерированию катион-радикалов, (алкил-)тиильных и полисульфидных радикалов, протона и атома водорода.
- Электрохимическая активация сероводорода, тиолов и сульфанов в реакциях с органическими соединениями
В связи с тем, что при электрохимическом окислении серосодержащих реагентов RSH (R=H, Alk, HSn) одномоментно или последовательно образуются разные активные интермедиаты, возможно целенаправленное проведение ряда реакций органических соединений с их участием: AdR, SR, S-циклизации, S-рециклизации, ди- и полимеризации.
Схему электрохимических превращений сероводорода, алкантиолов и сульфанов в присутствии органических соединений можно представить следующим образом:
R = H, С2H5, i-C3H7, n-C4H9, n-C6H13, HSn (n = 2-8) (5)
В зависимости от соотношения значений потенциалов окисления реагента RSH (R=H, Alk, HSn) и субстрата (А) предложенный способ электросинтеза органических соединений серы можно реализовать двумя путями: только при окислительной активации реагента (Епа(RSH) < Епа(А)) или при одновременной активации на аноде реагента и субстрата (Епа(RSH) >Епа(А)).
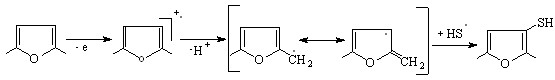
- Участие (алкил-)тиильных радикалов в реакциях с олефинами
Олефины линейного строения (гексен-1 Ib, гептен-1 IIb, октен-1 IIIb) окисляются при потенциалах > 2,3 В. В условиях анодного генерирования H2S+ (R = H, Alk) в реакции с олефинами рассматриваются два механизма (а, б) присоединения серосодержащего реагента к субстрату с учетом фрагментации образующегося катион-радикала:
![]()
![]()
а) 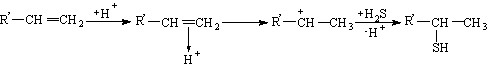
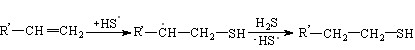
б) R’ = n-C4H9, n-C5H11, n-C6H13 (6)
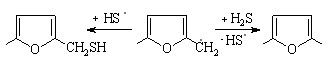
Образование алкантиолов-1 (выход по току 52-60 %) в ходе электросинтеза подтверждает радикальный механизм присоединении H2S к олефинам (рис. 4). ИК-спектр продуктов реакции свидетельствует о наличии валентных колебаний С–S-связи (670-770 см-1) и H–S-связи (800-940 см-1). Данные квантово-химических расчетов (РМ3 и ab initio в базисе 6-31G(d) GAMESS) подтверждают образование тиолов линейного строения, дальнейшее одноэлектронное окисление которых в условиях электросинтеза приводит к дисульфидам (выход по току 12-15 %):
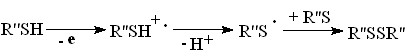
R” = n-C6H13, n-C7H15, n-C8H17 (7)
После электролиза (2 ч) смеси (H2S + Ib) молярное соотношение тиола и дисульфида (выход по току 20 %) составляет 2:1. Снижение молярного соотношения n-C6H13SH:(C6H13)2S2 (1,6:1) через 4 ч объясняется окислением тиола при потенциале проведения электролиза.
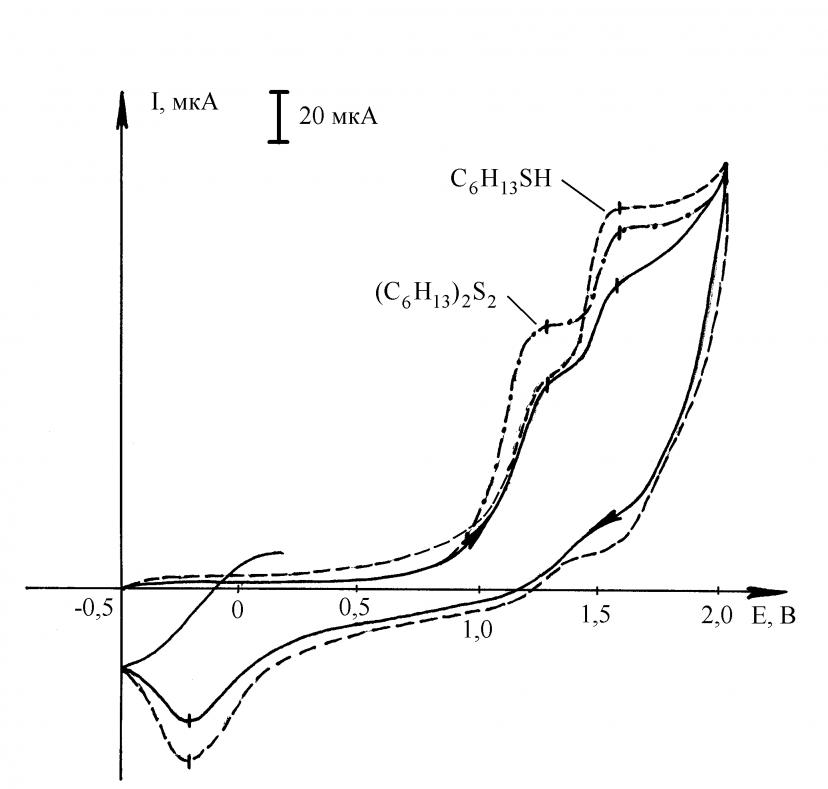 Рис. 4 – ЦВА смеси продуктов реакции H2S с гексеном-1 ____; идентификация добавкой стандартов (С=210-3 моль/л): n-C6H13SH_ _ _ ; (C6H13)2S2_._._. (CH3CN, Ag/AgCl, NBu4ClO4, Pt-анод, =4 ч, v(H2S)=2 л/ч) Рис. 4 – ЦВА смеси продуктов реакции H2S с гексеном-1 ____; идентификация добавкой стандартов (С=210-3 моль/л): n-C6H13SH_ _ _ ; (C6H13)2S2_._._. (CH3CN, Ag/AgCl, NBu4ClO4, Pt-анод, =4 ч, v(H2S)=2 л/ч) | При увеличении продолжительности электролиза наблюдается возрастание выхода по току (C6H13)2S2 до 28,9 %, а также образование сульфида по двум параллельным реакциям (присоединение активированного тиола к олефину и деструкция дисульфидов): |
Электролиз (6 ч) смеси (H2S + Ib) позволяет получить n-C6H13SH (42 %), (C6H13)2S (12 %), (C6H13)2S2 (21 %). Реакции H2S с олефинами IIb, IIIb протекают с образованием аналогичных соединений с соизмеримыми выходами по току. Обработка смеси продуктов реакции LiAlH4 приводит к восстановлению диалкилдисульфидов и увеличивает выход тиолов до 72-78%.
2.2 SH-, SR-функционализация ароматических углеводородов
в присутствии активированного сероводорода
Прямое тиолирование бензола и его производных возможно в жестких условиях. При взаимодействии H2S с бензолом в реакторе с неподвижным (движущимся) слоем угольного катализатора (500900 °С) выход тиофенола 2-5 %[2]
. Целесообразно было рассмотреть возможность прямого введения SH-группы в кольцо ароматического соединения посредством электрохимической активации H2S.
В качестве ароматических субстратов рассмотрены бензол Ic и его производные: a) соединения IIc-IXc и нитробензол XIIc (Епа > 2,0 В); б) анилин Xc (Епа < 1,6 В); в) 2,6-ди-трет.бутилфенол XIc (Епа = 1,6 В). При проведении электролиза смеси (H2S + Ic-IXc) в потенциостатическом режиме при потенциале окисления реагента (2 ч) получены ароматические тиолы (выход по току 75-80 %). Выход тиолов не зависит от природы заместителя в субстрате, что свидетельствует в пользу радикального механизма тиозамещения:
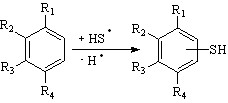
Ic-IXc
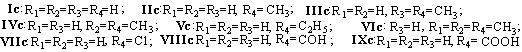 (10)
(10)
Хроматографический анализ продуктов реакции H2S с толуолом подтвердил образование смеси о-, м-, п-тиокрезолов и продукта тиозамещения атома водорода в метильной группе субстрата в соотношении 1,3:1,0:1,5:1,7. Выход по току продуктов тиолирования ароматических углеводородов (< 100%) указывает на отсутствие цепного характера радикальной реакции, что объясняется высокой активностью тиильных радикалов и одноэлектронным окислением ароматических тиолов при потенциале электролиза. При увеличении времени взаимодействия H2S с соединениями Ic-VIc образуются также дисульфиды и сульфиды:
 |
Ic-VIc
 (11)
(11)
В реакции H2S с анилином (1,0 В) выход продуктов тиолирования субстрата снижается из-за параллельно протекающей стадии окисления Xс при потенциале электролиза:
 (12)
(12)
Xс
SH-функционализация 2,6-ди-трет-бутилфенола (1,5 В) имеет большое значение благодаря особенностям продукта тиолирования: высокой антиоксидантной активности, способности к обратимому окислению, ингибированию цепных радикальных реакций и комплексообразованию с ионами металлов (Fe2+, Cu2+). Потенциал окисления пространственно-затрудненнных фенолов Епа(H2S), что позволяет в избытке реагента снизить энергозатраты на проведение электролиза и увеличить выход продукта реакции:

XIc (13)
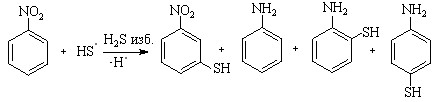
Электрохимическая реакция H2S (изб.) с нитробензолом приводит к изомерным продуктам SH-функционализации субстрата и анилину:
XIIc (14)
Превращение ароматического нитросоединения в амин протекает за счет генерирования протона при фрагментации катион-радикала сероводорода.
Таким образом, впервые удалось провести реакцию тиолирования ароматических соединений Iс-XIIc активированным H2S при 25 °С. Доказан радикальный механизм взаимодействия H2S с ароматическими субстратами при анодном окислении реагента.
2.3 Катион-радикал сероводорода в реакциях с замещенными
1,5-дикарбонильными соединениями
Возможность S-циклизации 1,5-дикарбонильных соединений в присутствии электрохимически окисленного H2S была рассмотрена для двух групп субстратов, отличающихся способностью к окислению. К первой группе относятся (алкил-) арилзамещенные 1,5-дикарбонильные соединения Id-Xd (Епа 2,0 В):
 Id-VIIId Id-VIIId | Id: R1=R5=C6H5, R2=R3=R4=H (2,36 В) IId: R1=R5=C6H5, R3=n-C6H4Cl, R2=R4=H (2,54 В) IIId: R1=R5=n-C6H4OCH3, R2=R3=R4=H (2,20 В) IVd: R1=R3=R5=C6H5, R2=H, R4=CH3 (2,32 В) Vd: R1=R3=R5=n-C6H4OCH3, R2=R4=H (1,82 В) VId: R1=R5=C6H5Cl, R2=R4=H (2,24 В) VIId: R1=R3=R5=C6H5, R2=R4=H (2,46 В) VIIId: R1=R5=o-, n-C6H4OCH3, R3=C6H5, R2=R4=H (1,82 В; 2,20 В) | 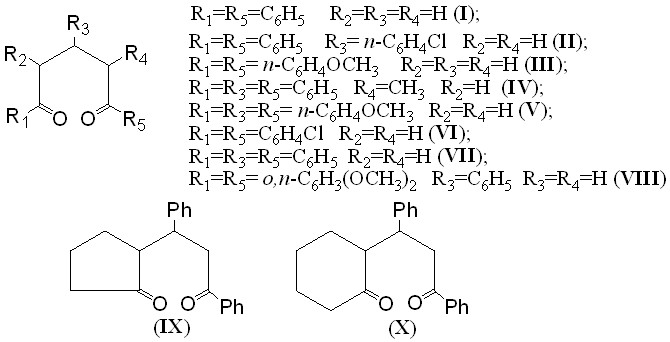 IXd (2,34 В) IXd (2,34 В)  Xd (2,38 В) Xd (2,38 В) |
В результате окислительной активации H2S при взаимодействии с соединениями Id-Xd образуются замещенные тиопираны (Епа = 1,5-1,7 В) без использования сильных кислот с выходом по току 50-62%. Реакция начинается со стадии протонирования карбонильной группы субстратов, при этом донором протона выступает катион-радикал H2S+:
 (15)
(15)
Id- Xd
Тиопираны в условиях электролиза окисляются до тиопирилиевых катионов (Епк = -0,28 – (-0,5) В, выход по току 27-35%) по механизму ЕСЕ, что можно представить на примере продукта S-циклизации 1,3-дифенил-3-(2-оксоциклопентил)-1-пропанона IХd схемой:
 (16)
(16)
Образование тиопиранильных радикалов подтверждено восстановлением тиопиранов на цинковом зеркале в резонаторе ЭПР-спектрометра. Так, ЭПР-спектр 2,4-дифенил-6Н-циклопента[b]тиопиранильного радикала (рис. 5) представляет собой триплет (1:2:1), обусловленный взаимодействием энергетических уровней неспаренного электрона с магнитными ядрами двух протонов в положении 7 гидридрированного кольца (ан = 0,69 мТл).
![ЭПР-спектр 2,4-дифенил-6Н-циклопента[b]тиопиранильного радикала-37](/images1/306473/epr-spektr-4-difenil-6n-ciklopenta.png)
Рис. 5 – ЭПР-спектр 2,4-дифенил-6Н-циклопента[b]тиопиранильного радикала (ТГФ, 298 К)
Каждая компонента триплета расщеплена в сложный мультиплет, представляющий собой триплет (1:2:1 с ан=23 мТл) септетов (1:6:15:20:15:6:1 с ан(Ph)= 0,08 мТл).
Для продуктов реакции активированного H2S с 1,3-дифенил-3-(2-оксоциклопентил)- IХd и 1,3-дифенил-3-(2-оксоциклогексил)-1-пропаноном Хd характерны дальнейшие превращения в условиях электролиза. На первой стадии окисления (2,4-дифенил-6Н-циклопента[b]-) 2,4-дифенил-6Н-циклогекса[b]тиопиранов (1,04 В; 1,20 В) образуются катион-радикалы, кислотные свойства которых усиливаются (рК 8), и далее – тиопирилиевые катионы.
Редокс-превращения 2,4-дифенил-6Н-циклопента[b] тиопирилиевого катиона (Епк = -0,38 В) описываются схемой:
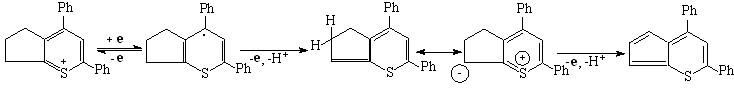
А В (17)
В этом случае образуется нестабильное ангидрооснование А и неустойчивая ароматическая структура – 2,4-дифенил-6Н-циклопента[b]тиопиранилиден В, который не удается выделить препаративно. Переход ангидрооснования А в соединение В сопряжен с отщеплением электрона и протона, что предполагает образование кратной связи в алицикле. Редокс-превращения 2,4-дифенил-6Н-циклогекса[b]тиопирилиевого катиона (Епк = -0,42 В) приводят к устойчивому ангидрооснованию С, что подтверждено при действии пиридина. Далее образуется димер D – 6,6’,7,7’-тетрагидро-2,2’,4,4’– тетрафенил-8,8’-би-5H-1-бензотиопиран:

С
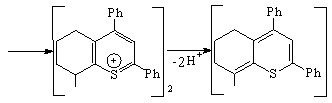 D (18)
D (18)
Электрохимическое окисление стабильного димера D в отличие от соединения В является обратимым, что согласуется с возможностью его препаративного выделения. Результаты анализа превращений тиопиранов методом ЦВА подтверждаются данными квантово-химических расчетов (метод ССП МО ЛКАО в базисе SТО-3G). Так, для двух путей превращений катиона 2,4-дифенилциклопента[b]тиопирилия (с образованием соединения В или димерного продукта) тепловые эффекты Е, соответственно, равны 1197,4 и 2170,8 кДж/моль.
Ко второй группе субстратов относятся халькогенбисцикланоны ХId-ХVd (ПИ = 9,30-9,42 эВ), которые окисляются при низких потенциалах (1,21,4 В) и обладают высокой фунгистатической активностью:
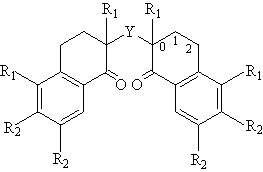 | Y = S, R1 = H, R2 = H XId (Епа= 1,32В); Y = S, R1=H, R2 = СH3 XIId (Епа= 1,26В); Y = S, R1=Br, R2 = СH3 XIIId (Епа= 1,40В); Y = Se, R1=H, R2 = H XIVd (Епа=1,24В); |  XVd (Епа=1,20В) XVd (Епа=1,20В) |
Исследуемые соединения можно расположить по их способности к окислению: (XVd)>(XIVd)>(XIId)>(XId)>(XIIId). По данным квантово-химических расчетов первичный катион-радикальный центр локализован преимущественно на гетероатоме. Для бисцикланонов ХId-ХVd положение уходящего протона (0-2) при фрагментации катион-радикалов зависит от их строения. Значения тепловых эффектов стадии депротонирования лежат в диапазоне 591,7-720,1 кДж/моль.
Окислительное инициирование реакции H2S с соединениями ХId-ХVd на аноде приводит к продуктам редокс-превращений субстратов. Электрохимические реакции халькогенбисцикланонов XId-XIIId на примере бис(1-оксо-1,2,3,4-тетрагидронафтил-2)сульфида XId описываются схемой:
![]()
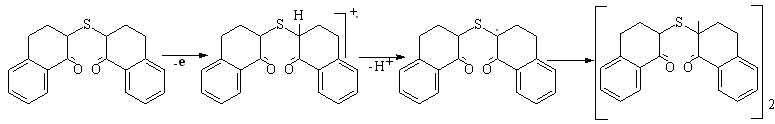 (19)
(19)
Димеры получены в независимом эксперименте при окислении 30%-ной Н2О2. В случае соединения XIIId димеризация радикалов протекает по положению 2 в отличие от бисцикланонов ХId, ХIId. Замена атома серы на селен приводит к снижению потенциала окисления бис(1-оксо-1,2,3,4-тетрагидронафтил-2)селенида XIVd (ПИ = 9,14 эВ), которое протекает с разрывом лабильной C–Se-связи:
![]()
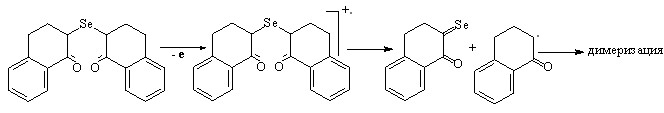 (20)
(20)
Необратимое окисление бис(борнеонил-2)сульфида XVd (ПИ = 9,10 эВ) протекает легче вследствие большего электронодонорного влияния алициклического фрагмента в сравнении с тетрагидронафталиновым:
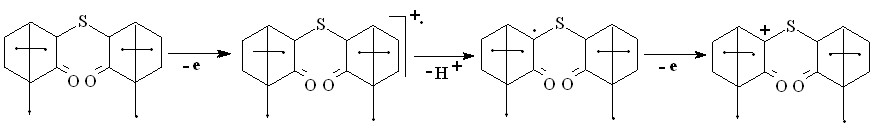 (21)
(21)
Проведенные исследования показали, что возможность S-циклизации замещеных 1,5-дикарбонильных соединений в тиопираны в условиях электрохимического окисления H2S определяется способностью исходных субстратов к окислению по сравнению с реагентом.
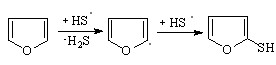
- Рециклизация пяти- и шестичленных O-, N-гетероциклов в присутствии электрохимически активированного сероводорода
Рециклизацию фурана (пиррола) в тиофен в присутствии H2S при 350-450 С проводят на Al2O3, модифицированном оксидами металлов (Ni, Cr, Co)[3]. Снижение энергозатрат (25 - 50 С) при замене атомов кислорода и азота в замещенном цикле на серу возможно в сильнокислой среде[4]
. Однако, в случае фурана (Епа = 2,0 В) и пиррола (Епа = 1,40 В) способ неприемлем ввиду их кислотной полимеризации.
В рамках разработанного подхода к окислительной активации H2S была рассмотрена S-рециклизация незамещенных фурана (пиррола). Электролиз смеси (H2S+фуран) в потенциостатическом режиме при потенциале окисления реагента (2 ч) приводит к тиофену (выход по току 34%), 2-тиофентиолу (выход по току 13 %) и смеси олиготиофенов. На первой стадии рециклизации в условиях окислительной активации H2S до катион-радикала образуется фураниевый катион:
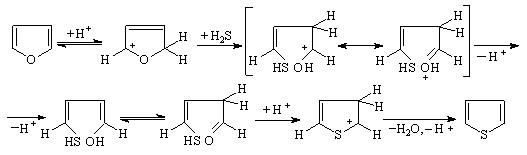 (22)
(22)
При рециклизации фурана выделяется Н2О, что способствует гидролитическому расщеплению гетероцикла, образованию 1,4-дикарбонильного соединения и его циклизации на серу:
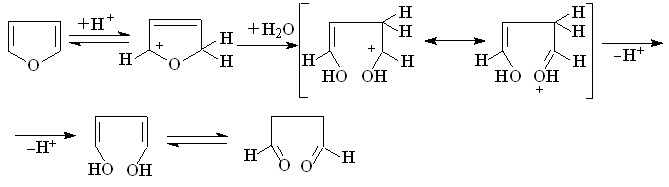
 (23)
(23)
 (24)
(24)
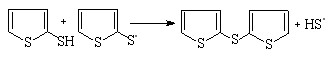
Превращение тиофена в 2-тиофентиол протекает по схеме:
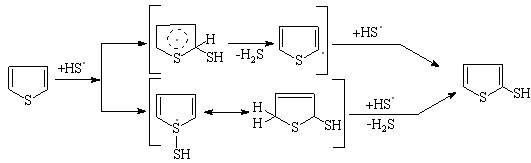 (25)
(25)
Выделение водорода за счет перенапряжения на катоде и генерирование тиильных радикалов способствует регенерации H2S. При увеличении времени электролиза (4 ч) протекает одноэлектронное окисление 2-тиофентиола c последующим образованием бис(2-тиенил)дисульфида (выход по току 28%):
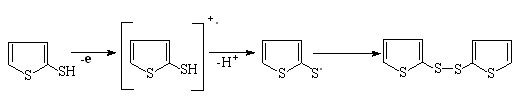 (26)
(26)
Бис(2-тиенил)сульфид (выход по току 3%) получен за счет генерирования неустойчивого 2-тиенилдисульфана и его диспропорционированию:
 (27)
(27)
Низкий выход бис(2-тиенил)сульфида обусловлен его превращением в 2-тиофентиол:
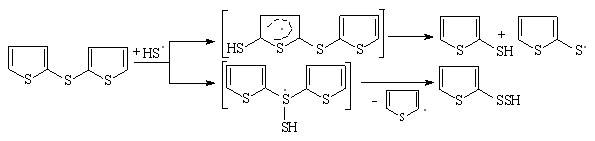 (28)
(28)
Увеличение времени электролиза (4 ч) способствует регенерации 2-тиофентиола (выход по току 20 %):
 (29)
(29)
Таутомерия 2-тиофентиола с последующим автотиилированием на воздухе приводит к 4-(2-тиенилтио-)тетрагидротиофен-2-тиону (выход по току до 10%):
 (30)
(30)
В инертной и аэробной атмосфере образуются тиофен (32-34 %) и 2-тиофентиол (8-12 %) с соизмеримым выходом по току. Снижение выхода по току тиофена (26%) и 2-тиофентиола (3%) в СН2Cl2 объясняется его низкой диэлектрической проницаемостью по сравнению с СН3СN. Более высокая вязкость и плотность дихлорметана затрудняют димеризацию и рекомбинацию 2-тиенилтиильных радикалов.
Кинетический контроль (рис. 6) реакции H2S с фураном методом ЦВА свидетельствует о псевдопервом порядке реакции (k=2,3710-4 с-1). Для подтверждения механизма SR в тиофене рассмотрено электрохимическое инициирование реакции H2S с 2,3,4,5-тетрабромтиофеном (2 ч), приводящее к (2-тио-)- и 2,5-дитио-3,4-дибромтиофену (молярное соотношение 1:1,8).
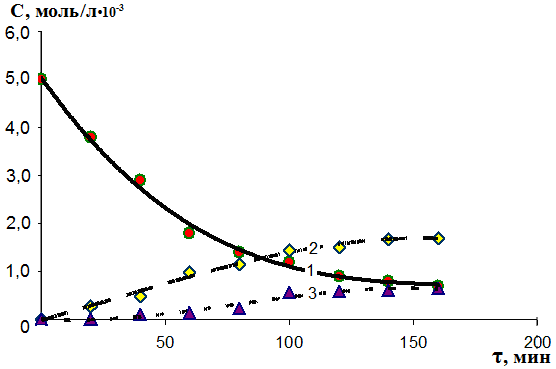 Рис. 6 – Кинетические кривые S-рециклизации фурана (1), образования тиофена (2) и 2-тиофентиола (3) в присутствии электрохимически активированного H2S. Рис. 6 – Кинетические кривые S-рециклизации фурана (1), образования тиофена (2) и 2-тиофентиола (3) в присутствии электрохимически активированного H2S. | 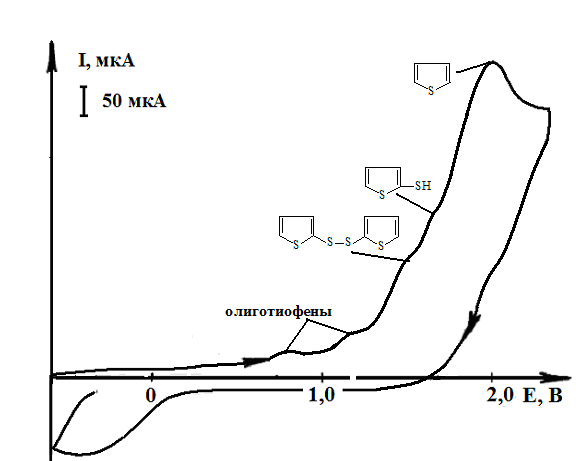 Рис. 7 – ЦВА смеси продуктов электролиза (H2S+фуран) (=6 ч, t=250C, СH3CN, Pt-анод, Ag/AgCl, 0,1 М NBu4ClO4, (H2S) = 2-3 л/ч) Рис. 7 – ЦВА смеси продуктов электролиза (H2S+фуран) (=6 ч, t=250C, СH3CN, Pt-анод, Ag/AgCl, 0,1 М NBu4ClO4, (H2S) = 2-3 л/ч) |
Препаративный электролиз смеси (H2S+фуран) (6 ч) при потенциале окисления реагента позволил получить тиофен, 2-тиофентиол и бис(2-тиенил)дисульфид с выходом по току 49,3%, 6,8% и 8,6%, соответственно, и олиготиофены (рис. 7). Степень превращения фурана в тиофен и его производные составляет 84,2%; конверсия тиофена в 2-тиофентиол, бис(2-тиенил)дисульфид и политиофены достигает 34,7%. Стадия протонирования при S-рециклизации фурана (PM3) термодинамически вероятней (на 101,42 кДж/моль), чем радикальное тиозамещение в О-гетероцикле. Наличие следовых количеств 2-фурантиола объясняется легкостью его рециклизации в S-содержащий аналог по сравнению с фураном (на 5,26 кДж/моль):
.  (31)
(31)
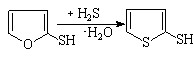 (32)
(32)
Таким образом, при окислительной активации H2S в реакции с фураном участвуют как ионы (протон, катион фурания), так и тиильный радикал, что приводит к широкому спектру серосодержащих гетероциклических соединений.
Для оценки влияния алкильных групп на способность O-гетероцикла к S-рециклизации в качестве субстрата был выбран 2,5-диметилфуран (1,46 В). В условиях электролиза смеси (H2S+2,5-диметилфуран) в безводных СН3СN, СН2Сl2 провести рециклизацию не удалось. На ЦВА наблюдается образование продуктов тиозамещения в цикле и в метильной группе 2,5-диметилфурана с выходом по току 8,7% и 13,3%:
 (33)
(33)
 (34)
(34)
Первой стадией превращений 2,5-диметилфурана в реакции с H2S является одноэлектронное окисление субстрата, причем в продуктах реакции фиксируются растворимые полимеры.
Учитывая способность замещенных фуранов к раскрытию цикла в присутствии нуклеофильного реагента (H2O) c образованием 1,4-дикарбонильных соединений, рассмотрено взаимодействие H2S с 2,5-диметилфураном в СН3СN при содержании воды (5-6%). По окончании электролиза (2 ч) образуется 2,5-диметилтиофен (выход по току 25%):
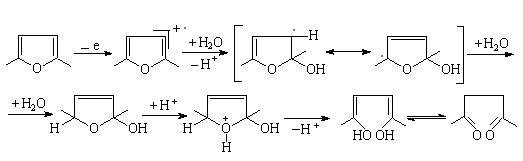 (35)
(35)
Введение заместителей в молекулу субстрата облегчает протонирование 2,5-диметилфурана по сравнению с фураном на 21 кДж/моль. В связи с этим можно учитывать и классический механизм рециклизации. Однако, замена атома кислорода на серу в 2,5-диметилфуране термодинамически менее выгодна, чем в фуране на 15,78 кДж/моль.
В условиях электрохимической активации Н2S в реакции с пирролом (2 ч) выход по току тиофена достигает 60%, при этом стадия протонирования пиррола протекает легче (на 58,12 кДж/моль по сравнению с фураном). Рециклизация пиррола (1,40 В) в присутствии Н2S происходит в условиях активации реагента и субстрата, что способствует повышению выхода тиофена:
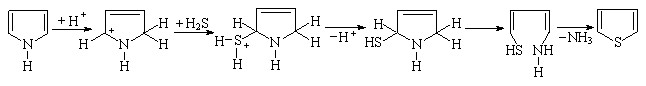 (36)
(36)
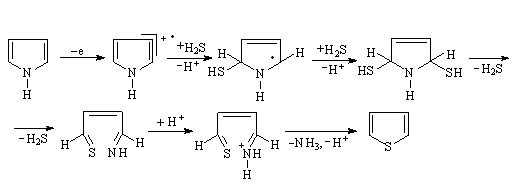 (37)
(37)
Окисление пиррола в условиях электросинтеза приводит к образованию нерастворимых полипиррольных пленок на поверхности анода. Рециклизация пиррола термодинамически более выгодна, чем тиозамещение в субстрате на 71,33 кДж/моль, что объясняет отсутствие в смеси продуктов реакции 2-пирролтиола.
Изучена возможность рециклизации ТГФ (Епа>2,40 В) в серосодержащий аналог с участием активированного H2S при 25 °С в отличие от высокотемпературного метода синтеза тиофана[5]
. Электролиз смеси (H2S+ТГФ) в CH3CN (2 ч) приводит к тиофану (выход по току 15%).
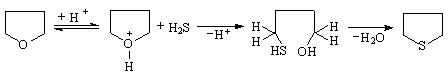 (38)
(38)
Увеличение времени электролиза (4 ч) повышает выход по току тиофана до 42 %. Рециклизация ТГФ по сравнению с фураном термодинамически менее вероятна (Н = 28,9 кДж/моль). Радикальное SH-замещения в ТГФ характеризуется более низким значением теплового эффекта по сравнению с S-рециклизацией (Н = 55,8 кДж/моль). Значения теплового эффекта тиолирования тиофана (Н = 9,6 кДж/моль) и реакции замещения в тиофене (Н = -33,2 кДж/моль) также свидетельствует об отсутствии изомерных тиофантиолов. Таким образом, нами обнаружена возможность получения тиофена на основе фурана, 2,5-диметилфурана, пиррола, а также тиофана из ТГФ в условиях активации сероводорода на аноде. Варьирование продолжительности электролиза позволяет синтезировать разные соединения тиофенового ряда.
- Реакции шестичленных O-, N-содержащих гетероциклов с электрохимически активированной формой сероводорода
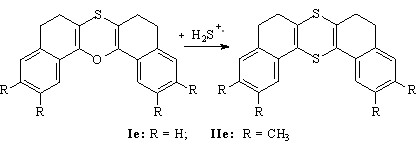
Для установления возможности рециклизации O-, N-содержащих шестичленных гетероциклов в присутствии окисленной формы H2S были выбраны оксатиины Ie-IIIe:
 | R = Н Iе (0,76; 1,28) R = СH3 IIе (0,72; 1,20) |  IIIe (1,32) IIIe (1,32) |
В резульатете электрохимической активации H2S при взаимодействии с соединениями Ie, IIe происходит окисление субстратов по механизму ЕЕС:
 (39)
(39)
Образование катион-радикалов оксатиинов Ie, IIe регистрируется в реакции с HClO4 методом УФ-спектроскопии ( = 470 нм). Дикатион реагирует с нейтральной молекулой оксатиина, что ведет к частичной регенерации гетероцикла. Превращения соединения IIIe в условиях электролиза с H2S протекают по механизму ЕСЕ:
 (40)
(40)
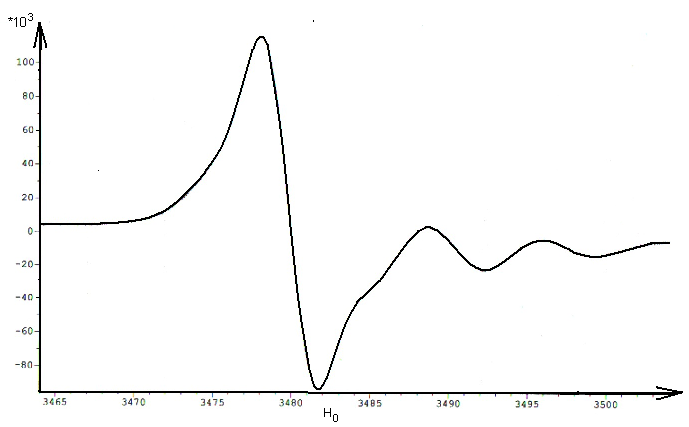 Рис. 8 – ЭПР-спектр, полученный при окислении соединения Ie серной кислотой (CH2Cl2, 293 К) Рис. 8 – ЭПР-спектр, полученный при окислении соединения Ie серной кислотой (CH2Cl2, 293 К) | Данные квантово-химического расчета путей превращений соединений Ie-IIIe подтверждают вероятность образования продукта, содержащего дополнительную двойную связь в алициклическом фрагменте (H = -25,8 – (-22,8) кДж/моль), в отличие от продукта димеризации по положениям 2-2’ (H = 5,1 - 19,9 кДж/моль). В спектре ЭПР (рис. 8) при взаимодействии соединения Ie с H2SO4 наблюдаются синглетный сигнал (g=2.00482), принадлежащий |
четвертичному атому углерода (s(C)=1/2; 1,11%), и триплетный сигнал на атоме серы (s(S)=3/2; 0,75%). Так как невозможно установить локализацию неспаренного электрона был сделан вывод об отсутствии стадии депротонирования катион-радикала соединения Ie.
Замена атома кислорода на серу при окислительной активации Н2S в реакции с соединениями Ie, IIe приводит к дитиинам E, F (выход по току 35 – 38 %):
 (41)
(41)
Ie, IIe E (R = Н), F (R = СH3)
Вероятно, S-рециклизация соединений Ie, IIe начинается со стадии протонирования атома кислорода. Потенциал окисления дитиинов E (0,82 В; 1,34 В), F (0,78 В; 1,26 В) смещается в положительную область (по сравнению с Епа субстрата) из-за более высокой их устойчивости ввиду равномерного распределения электронной плотности между двумя атомами серы. В случае реакции катион-радикала сероводорода с диазепином IIIe S-рециклизация субстрата не происходит.
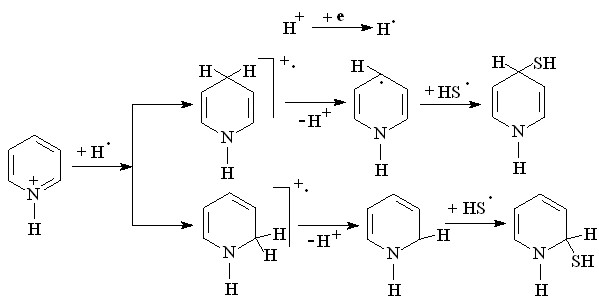
Целесообразным было рассмотреть возможность вовлечения активированного H2S во взаимодействие с пиридином Iа и его бензаннелированными аналогами IIa, IIIа для получения соединений с двумя реакционными центрами, которые являются основой для биологически активных веществ. Увеличение стабильности оснований Iа-IIIа при переводе их в катионную форму (G=7,3 7,7эВ, RHF в базисе 6-31G(d,p)) в присутствии катион-радикала H2S+. определяет направление превращений, которые на примере соединения Iа можно описать схемой:
 (42)
(42)
Увеличение концентрации H2S приводит к перенапряжению водорода на катоде, участвующего в реакции с катионами пиридиния, хинолиния и акридиния. В ходе электролиза смеси (соединение Iа-IIIа + H2S) при потенциале окисления реагента (2,5 ч) субстраты не подвергаются окислению. Продукты радикального тиоприсоединения (1,14 – 1,48 В) H2S к основаниям Iа-IIIа и протонированные формы субстратов образуются в соотношении 1:1. На ЭСП наблюдаются батохромные сдвиги максимумов поглощения продуктов относительно исходных соединений Iа-IIIа. Степень превращения соединений Iа-IIIа в тиопроизводные (7579 %) не зависит от природы субстратов. В препаративном электросинтезе на основе Н2S и соединения Iа (5 ч) образуется смесь изомеров – 4-меркапто-1,4-дигидропиридина и 2-меркапто-1,2-дигидропиридина (выход по току 23%) при соотношении 1,8:1.
Как следует из полученных данных, рециклизация оксатиинов протекает за счет атаки субстрата – продуктом фрагментации (H+) катион-радикала H2S. S-рециклизация пиридина и его конденсированных аналогов не идет ввиду устойчивости гетероцикличских катионов к раскрытию цикла и конечными продуктами реакции с сероводородом являются меркаптодигидропиридины.
2.6 Использование косвенного электрохимического способа активации сероводорода в синтезе органических соединений серы
В последнее время большой интерес у химиков-органиков вызывают как прямые, так и косвенные электрохимические способы активации молекул, направленные на снижение энергозатрат на их взаимодействие с органическими соединениями.
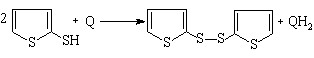
Для промотирования электросинтеза тиофена и его производных использован редокс-катализатор (3,5-ди-трет-бутил-о-бензохинон – Q), восстановленная форма (QH2) которого окисляется при более низком потенциале (1,2 В), чем H2S. Электрокаталитическая реакция H2S с фураном протекает при снижении энергозатрат (Е = Епа(H2S) - Епа(QH2) = 0,4 В).
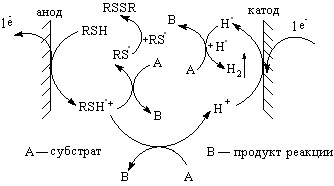
Принцип действия предложенной системы «анод-о-бензохинон-сероводород» в СН3СN возможно описать следующим образом:
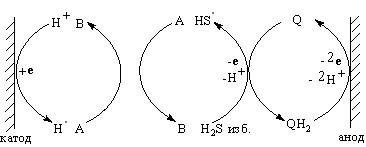
А – субстрат; B – продукт реакции (43)
Взаимодействие H2S с фураном (рис. 9) в присутствии Q (3 ч) приводит к тиофену (59%), 2-тиофентиолу (17,6%), бис(2-тиенил)дисульфиду (2,1%) и нерастворимым политиофенам.
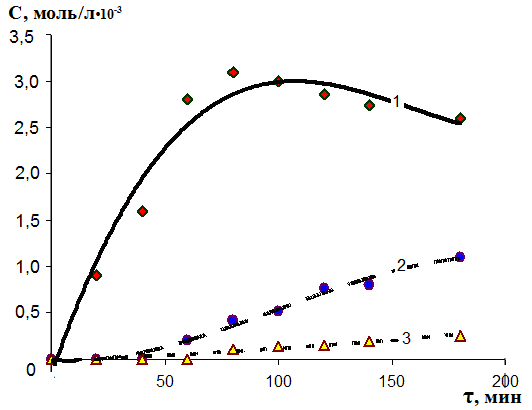 Рис. 9 – Кинетические кривые образования тиофена (1), тиофентиола (2), бис(2-тиенил)дисульфида (3) в условиях электрокатализа (=3 ч). Рис. 9 – Кинетические кривые образования тиофена (1), тиофентиола (2), бис(2-тиенил)дисульфида (3) в условиях электрокатализа (=3 ч). | Степень превращения фурана достигает 100%, чего не удается при проведении электролиза в отсутствии о-бензохинона. Увеличение времени электрокатализа (6 ч) приводит к повышению выхода бис(2-тиенил)дисульфида (22%). Таким образом, редокс-пара «Q – QH2» выполняет электрокаталитическую роль в реакции сероводорода с фураном и повышает эффективность рециклизации O-содержащего цикла в тиофеновый аналог. |
Для снижения энергозатрат при проведении электросинтеза органических соединений серы в CH3CN применялась также система «анод-основание-сероводород». Сущность данной комбинированной системы основана на депротонировании молекулы H2S основанием Льюиса и окислением тиолат-аниона на аноде (0,14 В):
H2S + :B HB+ + HS-
B=(C2H5)3N, (CH3)3N
HS- - е HS (44)
Взаимодействие H2S с (C2H5)3N (0,9 В), (CH3)3N (1,00 В) снижает потенциал электрогенерирования тиильных радикалов ( на 1,40 В) по сравнению с прямым окислением сероводорода (рис. 10). Система «анод-основание-сероводород» успешно использована нами для SH-функционализации органических соединений:
 (45)
(45)
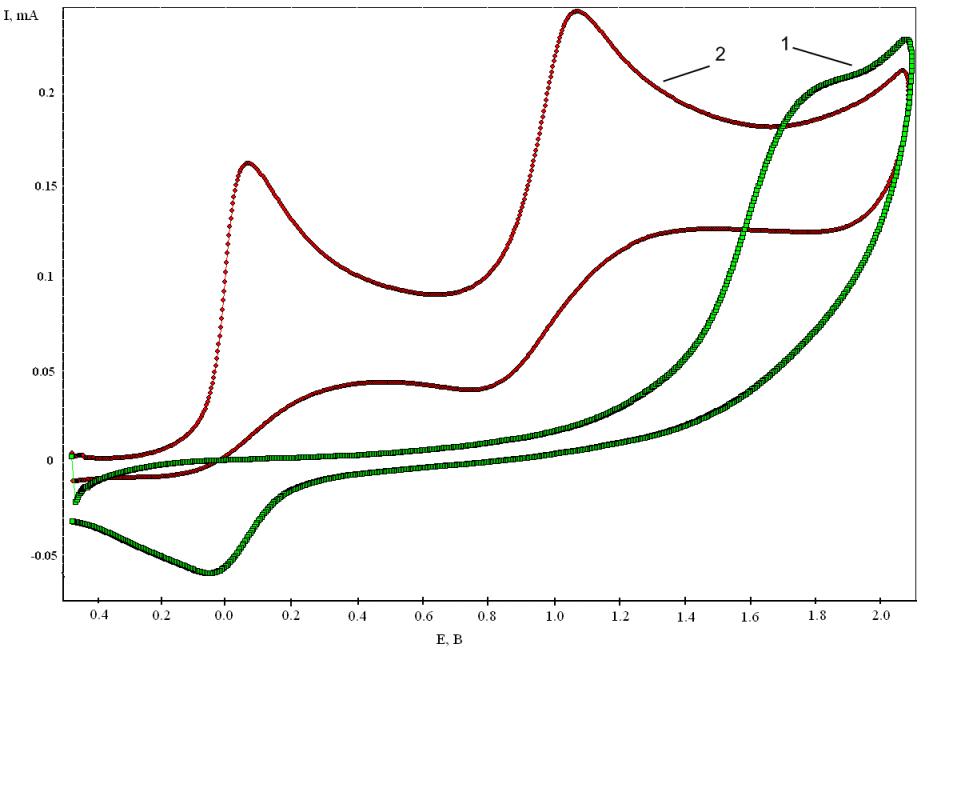 Рис. 10 – ЦВА 1) окисления H2S; 2) смеси H2S с (C2H5)3N (1:3) (C(H2S) = 510-2 моль/л, Pt-электрод, СН3СN, 0,1М NBu4ClO4, Ag/AgCl) Рис. 10 – ЦВА 1) окисления H2S; 2) смеси H2S с (C2H5)3N (1:3) (C(H2S) = 510-2 моль/л, Pt-электрод, СН3СN, 0,1М NBu4ClO4, Ag/AgCl) | Электросинтез органических производных серы на основе сероводорода в присутствии оснований Льюиса проведен при низком анодном потенциале (0, 14 В). Таким образом, показана эффективность окислительной активации H2S, тиолов и cульфанов на аноде прямым и косвенными способами. Преимуществом данного подхода к реализации реакций с участием RSH (R=H, Alk, HSn) является возможность управления элекросинтезом. Варьирование спектра органических соединений серы возможно за счет |
различной продолжительности реакции.
Однако электрохимический синтез органических соединений не всегда удобен. Так, в случае гетероциклических соединений электролиз сопровождается пассивацией поверхности анода ввиду их полимеризации. Снижение выхода продуктов реакции обусловлено многостадийностью их выделения из-за присутствия в реакционной смеси фонового электролита.
- Активация сероводорода и тиолов пространственно-затрудненными о-, п-бензохинонами в реакциях с органическими соединениями
С целью поиска альтернативного подхода к синтезу органических соединений серы через стадию одноэлектронного переноса были изучены превращения рассмотренных выше субстратов с H2S при использовании окислителей. В связи с этим предпринята замена анода на пространственно-затрудненные о- и п-бензохиноны (Q):
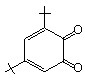 | 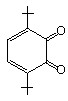 |  | 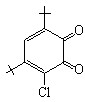 | 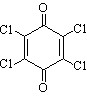 | 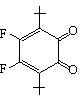 |  |
Q1 (-0,42 В) Q2 (-0,46 В) Q3 (-0,72 В) Q4 (-0,36 В) Q5 (-0,54 В) Q6 (-0,32 В) Q7 (-0,32 В)
Исследуемые окислители обратимо восстанавливаются в две одноэлектронные стадии до анион-радикала и дианиона. Активация RSH (R=H, Alk, HSn) в присутствии Q, как и при их электрохимическом окислении, ведет к образованию соответствующих катион-радикалов. Фрагментация способствует протонированию дианионов о- и п-бензохинонов с образованием замещенных пирокатехинов или гидрохинонов (QH2) при потенциалах 1,1 -1,4 В:
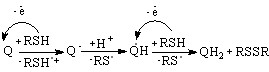 R=H, Alk, H2Sn (46)
R=H, Alk, H2Sn (46)
При окислении сероводорода и сульфанов о- и п-бензохинонами образуется молекулярная сера, в случае алкантиолов – дисульфиды. Активаторы Q1-Q7 регенерируются кислородом воздуха, что зависит от положения и природы заместителей:
![]() (47)
(47)
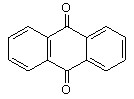
Наиболее эффективными окислителями Н2S оказались о-бензохиноны Q1, Q4, обладающие оптимальной скоростью регенерации (24 – 48 ч) и числом рабочих циклов – 14 и 11, соответственно. Трет-бутильные заместители в о-бензохинонах затрудняют, а атомы галогенов облегчают перенос электрона от молекулы H2S. Антрахинон обладает низкой каталитической активностью, так как восстанавливается при высоком значении потенциала и требует более длительного времени регенерации (7 сут).
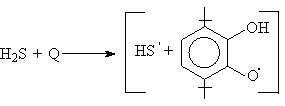
Два альтернативных процесса – перенос атома водорода от молекулы Н2S к молекуле Q или протона на анион-радикал бензохинона – приводят к образованию ион-радикальной пары. Окисление Н2S в присутствии Q2 (молярное соотношение 1:1) методом ЭПР приводит к генерированию 3,6-ди-трет-бутил-2-гидроксифеноксильного радикала (рис.11а), сигнал которого возрастает из-за образования о-гидрохинона и диспропорционирования (рис. 11б). При облучении смеси Н2S и Q2 при 77 К (рис.12) регистрируется радикальная пара, стабильная в твердой фазе (тензор тонкой структуры PhCО = 42,5 э):
 (48)
(48)
Катион-радикал Н2S и анион-радикал 3,6-ди-трет-бутил-семихинолят-аниона не фиксируются из-за высокой кислотности и основности частиц.
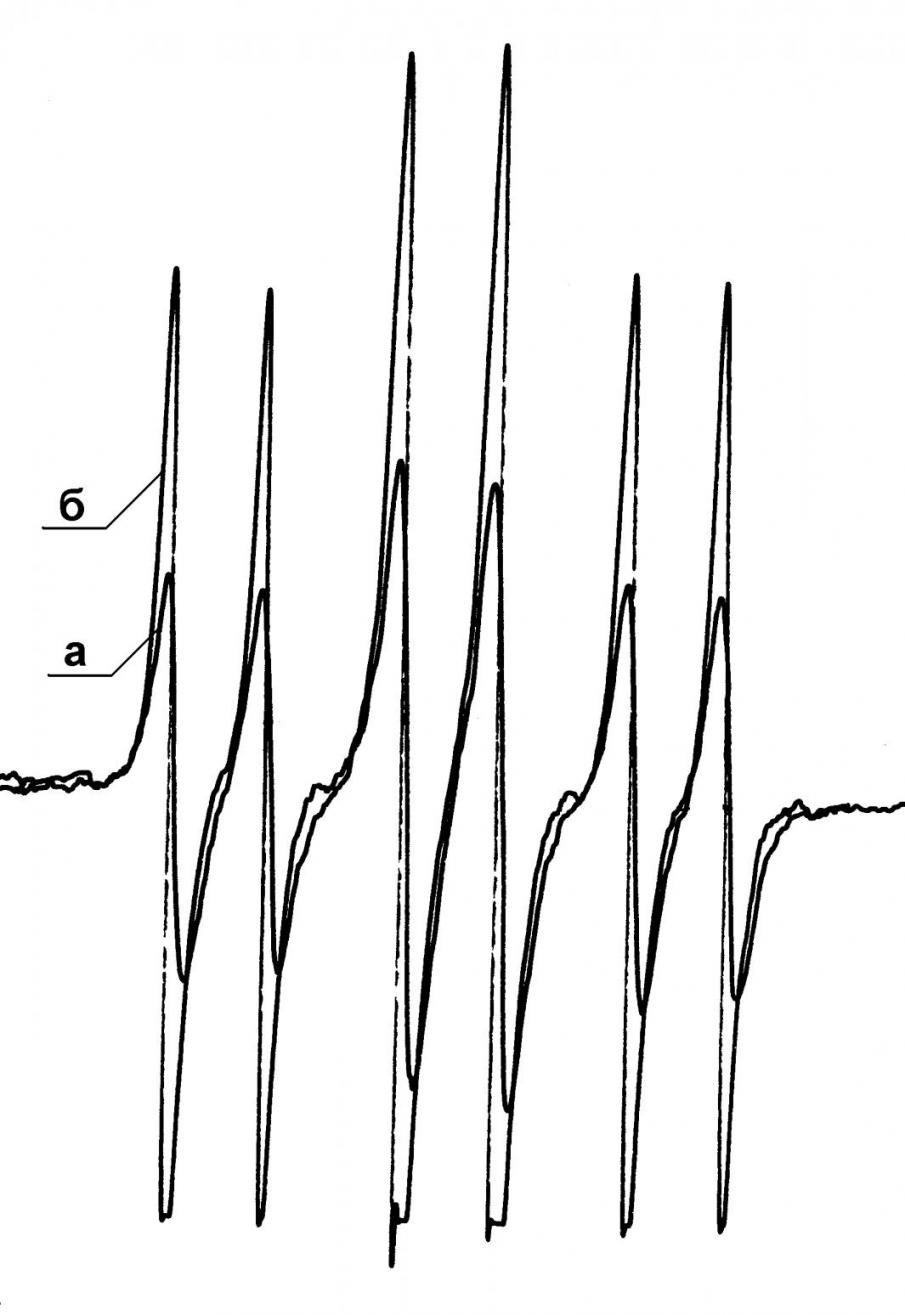 Рис. 11 – Спектр ЭПР продукта взаимодействия Н2S с 3,6-ди-трет-бутил-о-бензохиноном : а) в начальный момент реакции; б) через 10-15 мин (толуол, 25°С) Рис. 11 – Спектр ЭПР продукта взаимодействия Н2S с 3,6-ди-трет-бутил-о-бензохиноном : а) в начальный момент реакции; б) через 10-15 мин (толуол, 25°С) | 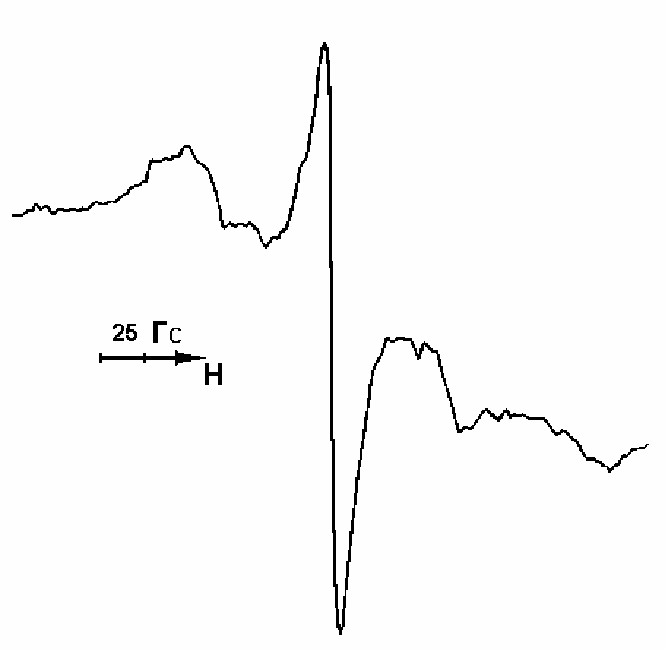 Рис. 12 - Спектр ЭПР продукта ваимодействия Н2S с 3,6-ди-трет-бутил-о-бензохиноном (толуол, 77 К). |
В связи с доступностью о-бензохинонов и рядом их преимуществ интересно было изучить реакции H2S с органическими соединениями в присутствии окислителей Q1, Q4. Для активации сероводорода в реакции с гексеном-1 (рис. 13) использовали окислитель Q1 (0,1% масс). При постоянном токе H2S (1 - 2 л/ч) образуются n-С6Н13SH и (С6Н13)2S (молярное соотношение 2,1:1), cодержание которых завист от продолжительности реакции (рис. 14).
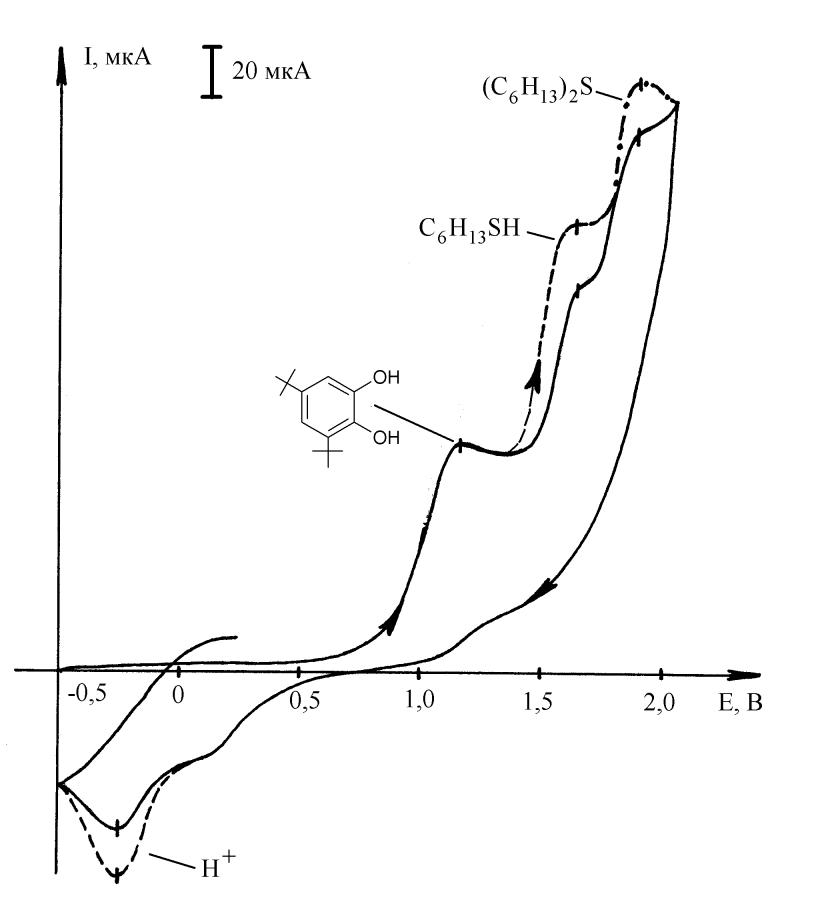 Рис. 13 – ЦВА смеси продуктов реакции H2S с гексеном-1 в присутствии Q1___; идентификация добавкой стандартов (С=210-3 моль/л): _ _ _ n-С6Н13SH; _._._ (С6Н13)2S (t=25°C, СН3СN, Pt-электрод, Ag/AgCl, 0,1 М NBu4ClO4) Рис. 13 – ЦВА смеси продуктов реакции H2S с гексеном-1 в присутствии Q1___; идентификация добавкой стандартов (С=210-3 моль/л): _ _ _ n-С6Н13SH; _._._ (С6Н13)2S (t=25°C, СН3СN, Pt-электрод, Ag/AgCl, 0,1 М NBu4ClO4) | Окисление гексантиола-1 в присутствии Q1 приводит к катион-радикалу и его фрагментации с отрывом протона. Отсутствие в продуктах реакции (С6Н13)2S2, фиксируемого при электрохимическом инициировании реакции H2S с гексеном-1, объясняется восстановлением дисульфида до тиола в присутствии Q1H: 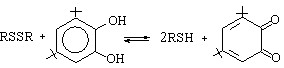 R=С6H13 (49) R=С6H13 (49) |
| При проведении препаративного синтеза органических соединений серы варьировали содержание окислителя и соотношение H2S:субстрат. Увеличение скорости подачи H2S до 2-3 л/ч в реакции с гексеном-1 способствует повышению выхода n-С6Н13SH. Повышение концентрации Q1 способствует росту селективности реакции присоединения тиола к олефину. Оптимальные условия реакции H2S с олефинами: время – 3 ч, | 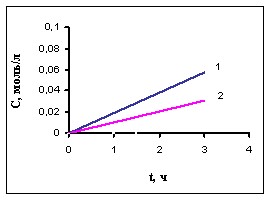 , ч Рис. 14 - Зависимость концентрации продуктов реакции H2S с гексеном-1 от времени: 1 – n-С6Н13SH, 2 - (С6Н13)2S ((H2S)=1-2 л/ч; =3 ч, С(Q1)=510-3 моль/л, t=25°C) , ч Рис. 14 - Зависимость концентрации продуктов реакции H2S с гексеном-1 от времени: 1 – n-С6Н13SH, 2 - (С6Н13)2S ((H2S)=1-2 л/ч; =3 ч, С(Q1)=510-3 моль/л, t=25°C) |
скорость подачи реагента – 2-3 л/ч, содержание окислителя – 0,2% масс. В случае реакции сероводорода с гексеном-1 получены гексантиол-1 (32%) и дигексилсульфид (молярное соотношение 3,3:1).
Для окисления алкантиолов в присутствии Q (рис. 15) установлена закономерность: увеличение длины углеводородного радикала в молекуле тиола замедляет скорость его окисления.
| Кинетический расчет свидетельствуют о бимолекулярности реакции тиолов с окислителями. При проведении реакции H2S c ароматическими углеводородами Ic, IIc, IXc в присутствии Q1-Q3, Q5 получены соответствующие тиофенолы (табл. 2), скорость образования которых зависит от природы субстрата и окислителя. |  , мин Рис. 15 - Кинетические кривые окисления алкантиолов в присутствии Q1 (1:1), 250C , мин Рис. 15 - Кинетические кривые окисления алкантиолов в присутствии Q1 (1:1), 250C | |||||||||||||||||||||||||||||||||||||||||||
Табл. 2 Результаты реакций H2S с ароматическими соединениями с в присутствии Q1-Q4 (60 мин)*, t=25°C
Примечание: * Саром. субстрата = Схинона = 510–3 моль/л; ** скорость образования тиофенолов, выраженная в моль/мин продукта по отношению к количеству Q | Реакция H2S с 1,5-дикарбонильными соединениями Id, VIId, IXd при использовании Q1 протекает с образованием тиопиранов (5865%) и солей тиопирилия (2023%). Как и ожидалось, активация H2S в бензохинонами также приводит к рециклизации O-, N-гетероциков в тиофен. При взаимодействии H2S с фураном (3 ч) в присутствии Q1 (0,1% масс.) образуются тиофен (23,5%), 2-тиофентиол (3,4%) и бис(2-тиенил)дисульфид (1,3%) (рис. 16). | |||||||||||||||||||||||||||||||||||||||||||
Выход тиофена и его производных удалось увеличить в 2,5-3 раза за счет регенерации окислителей кислородом воздуха. В масс-спектрах продуктов реакции H2S с фураном фиксируются молекулярные ионы m/z: 116 [М]+, 230 [М]+, отвечающие 2-тиофентиолу и бис(2-тиенил)дисульфиду. Ион m/z: 100 [М]+ соответствует 2-фурантиолу, полученному в следовых количествах. При увеличении времени реакции (4 ч) образуется бис(2-тиенил)сульфид (молекулярный ион m/z: 198 [М]+): |
Рис. 16 – ЦВА смеси продуктов реакции H2S с фураном в присутствии Q1 (СН3СN, Pt-электрод, Ag/AgCl, 0,1 М NBu4ClO4, =3 ч, 250C) | |||||||||||||||||||||||||||||||||||||||||||
 (50)
(50)
Наибольший выход тиофена (26,6 %), 2-тиофентиола (4,7%) и бис(2-тиенил)дисульфида (3,5%) достигается при использовании окислителя Q4. Природа окислителя влияет на вторичные превращения тиофена в 2-тиофентиол и бис(2-тиенил)дисульфид (табл. 3).
Табл. 3
Соотношение продуктов реакции H2S с фураном в присутствии Q (=3 ч, t=25°C)
Окислитель |  | ||
| Q1 | 1,5 | 1,1 | 2,0 |
| Q2 | 1,0 | 1,0 | 1,0 |
| Q4 | 1,7 | 1,4 | 4,0 |
Степень превращения фурана в серосодержащие гетероциклы в присутствии Q4 достигает 68 %, окислителя Q1 – 44%. С увеличением продолжительности (8 ч) реакции H2S с фураном способствует возрастанию выхода бис(2-тиенил)дисульфида (50 %) за счет регенерации окислителей:
 (51)
(51)
По аналогии с реакцией H2S и 2,3,4,5-тетрабромтиофена следовало рассмотреть воможность замены атома галогена в окислителях на тиильный радикал. Было изучено взаимодействие H2S с о-бензохинонами Q4, Q5, Q7 в условиях электролиза при потенциале окисления пирокатехинов QH.
Замещение атома хлора на HS· в активаторе Q4 не идет, очевидно, вследствие пространственного экранирования трет.-бутильными группами. Реакция H2S c окислителями Q5, Q7 приводит к продуктам тиозамещения в о-бензохинонах:
 (52)
(52)
При взаимодействии H2S с пирролом в присутствии Q1, Q2, Q4 продукт рециклизации субстрата не образуется. Вероятно, это связано с высокой окислительной способностью о-бензохинонов по отношению к пирролу, что приводит к образованию полипиррола. В реакции H2S с ТГФ в присутствии Q1 получен тиофан (3 ч ) с незначительным выходом (3%).
Реакция H2S с 2,5-диметилфураном в присутствии Q1 в CH3CN (5-6% H2O) протекает с образованием 2,5-диметилтиофена (49%). В безв.CH3CN взаимодействие не идет и вероятно, получен продукт присоединения 2,5-диметилфурана к о-бензохинону. Следовательно, о-бензохинон Q1 преимущественно выступает в роли реагента, атакующего гетероцикл, и не выполняет функцию окислителя сероводорода.
В последнее время для получения серосодержащих органических полимеров используют молекулярную серу. Так, эффективно протекает поликонденсация серы с ароматическими соединениями при 100-250 °С.
По аналогии было проведено взаимодействие H2S с фураном (3 ч) в присутствии Q1 и серы при 25 °С, приводящее к тиофену (22,4%) и 2-тиофентиолу (6,5%). Сера реагирует с активированным H2S, инициируя образование тиильных радикалов, концентрация которых определяет выход и природу серосодержащих гетероциклических соединений. Особенностью применения системы «сера-сероводород» является увеличение выхода политиофенов до 20-25%.
- Синтез серосодержащих органических соединений при активации сероводорода и тиолов в присутствии комплексов с редокс-лигандами
Редокс-активность хиноидных систем можно повысить за счет координации переходным металлом, что вызывает существенные изменения электронных, структурных свойств лигандов и влияет на их реакционную способность. Металло-промотируемый синтез, реализуемый благодаря превращениям лигандов, расширяет возможности предлагаемого подхода к окислительной активации RSH (R = H, Alk) и получению органических соединений серы в достаточно мягких условиях.
4.1 Синтез и электрохимические свойства комплексов
металлов VIII группы с peдокс- активными лигандами
Комплексы металлов VIII группы с peдокс-активными лигандами 1-21 (комплексы 10-12 синтезированы впервые) были получены реакцией неорганических солей металлов и органических лигандов в водно-ацетонитрильных растворах[6]
.
Реакция протекает через образование тетрааминных или сульфидных комплексов, окислительное дегидрирование которых приводит к образованию анион-радикальных форм лигандов и их координации ионом металла.
Методом ЦВА исследовано поведение моноядерных комплексов никеля, платины и палладия с N,N-, N,S-, S,S-координированными лигандами.
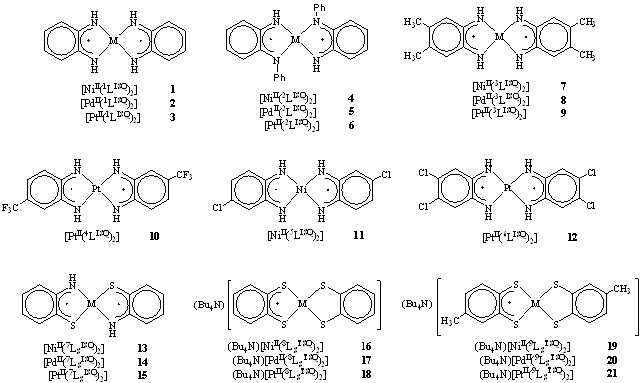
Установлена общая закономерность протекания редокс-процессов соединений 1-21 (рис. 17), приводящих к пяти бис-(хелатным)-металлокомплексам [MII–X,Y] Z (z=-2,-1,0,+1,+2):
![(53) ЦВА комплекса [PdII(2LISQ)2] (CH2Cl2:CH3CN (1:1), V =-104](/images1/306473/53-cva-kompleksa-pdii2lisq2-ch2c.jpg) (53)
(53)
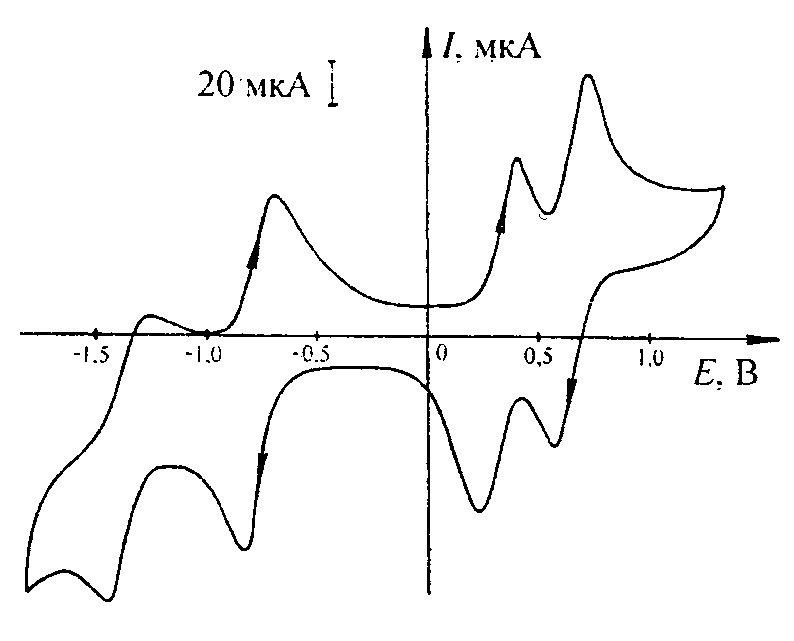 Рис. 17 – ЦВА комплекса [PdII(2LISQ)2] (CH2Cl2:CH3CN (1:1), V = 0.5 В/с, Ag/AgCl, С=510-3 моль/л, Ar, Pt-электрод, 0.1 М Bu4NClO4) Рис. 17 – ЦВА комплекса [PdII(2LISQ)2] (CH2Cl2:CH3CN (1:1), V = 0.5 В/с, Ag/AgCl, С=510-3 моль/л, Ar, Pt-электрод, 0.1 М Bu4NClO4) | Значения редокс-потенциалов комплексов 1-21 (табл. 4) достаточно близки для одного типа лигандов и зависят от природы заместителей в них – это подтверждает участие лигандов в редокс-превращениях. Семихинондииминовые комплексы 1-3 и их N-фенилзамещенные аналоги 4-6 имеют близкие значения редокс-потенциалов. В случае N-фенил-о-фенилендиамина энергия СЗМО в моноанионе снижается по сравнению с о-фенилендиамином, что объясняет смещение |
значения потенциала второго редокс-перехода в анодную область.
Табл. 4 Электрохимические характеристики моноядерных комплексов 1-21 ( = 200 мВ/с, Ag/AgCl, С=510-3 моль/л, Pt-электрод, 0.1 М Bu4NСlO4)
Примечание: а-ДМФА; b-MeCN:CH2Cl2=1:1; c-ТГФ:CH2Cl2=1:1; d-CH2Cl2; | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Введение акцепторных заместителей стабилизирует моноанионные формы и снижает устойчивость окисленных комплексов 10-12. Рассмотренный выше подход к SH-функционализации анилина в присутствии активированного H2S использовали в синтезе комплексов 13-15 с N,S-координированными лигандами. Потенциалы восстановления комплексов 13-15 смещены в анодную область по сравнению с комплексами 1-6, что объяснятся более эффективным перекрыванием молекулярных орбиталей атомов серы и металла. Высокие значения анодных потенциалов при низких значениях катодных для комплекса 15 определяют слабовыраженный семихиноидный характер лигандов, в которых электронная плотность локализована преимущественно на атомах серы.
Для дитиолатных комплексов 16-21 стабильными являются моноанионные формы с противоионом Bu4N+, которые не реагируют с H2S в отличие от их активных окисленных форм. Комплексы 16-21 восстанавливаются (окисляются) в две одноэлектронные обратимые стадии:
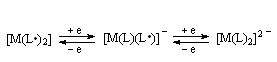 (54)
(54)
Комплексы 16-21 парамагнитны (S = 1/2), спектр ЭПР комплекса 16 в растворе (рис. 18) представляет собой синглет (gизо = 2.085) без видимой сверхтонкой структуры. В СH2Cl2 при 300 К также наблюдается синглет с сопоставимым значением ширины линии (H = 14 Гс, gизо= 2.082). Величина g-фактора отлична от значений, характерных для лиганд-центрированных радикалов (gизо = 2.000-2.004), что свидетельствует о значительном вкладе атомных орбиталей металла в СЗМО (до 35%). Это связано с влиянием спин-орбитального взаимодействия на значение g-фактора.
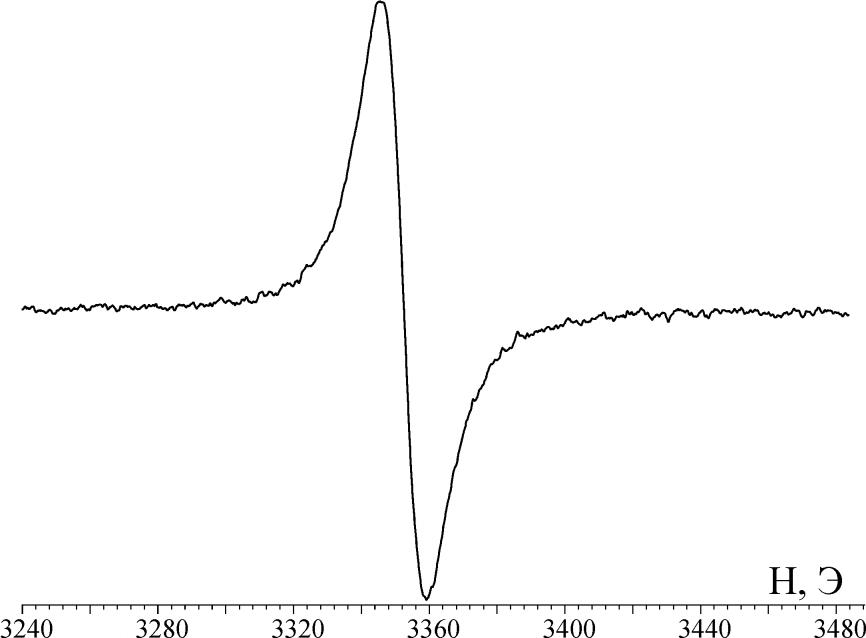 Рис. 18 – ЭПР-спектр комплекса [Bu4N][NiII(8 Рис. 18 – ЭПР-спектр комплекса [Bu4N][NiII(8 | В комплексах 1-15 происходит перенос заряда ( = 730-850 нм) между анион-радикальными о-семихинолятными лигандами. При одноэлектронном окислении нейтральных комплексов 16-21 исчезает максимум поглощения ( = 890-1100 нм), что коррелирует с поведением аналогичных моноанионных форм комплексов с N,N- и N,S-координированными семихинолятными лигандами. |
Комплекс никеля 16 представляет собой анион-радикальную соль, окисляющуюся до неустойчивой бирадикальной формы – время жизни составляет 15 мин при 298 К. В ходе электролиза соединения 16 на ЭСП наблюдается батохромный сдвиг (=700850 нм), свидетельствующий о превращении анион-радикальной формы комплекса в радикальную. Аналогичные превращения характерны и для комплекса 19.
Для оценки термодинамической стабильности о-семихинолятных комплексов Ni, Pd, Pt и установления влияния природы металла и лиганда на редокс-поведение комплексов металлов корректно использовать значения энергетической щели, которые в отличие от значений изменения свободной энергии Гиббса G, не зависят от условий измерения потенциалов, поскольку рассчитываются на основании величин полуволн обратимых одноэлектронных реакций:
Евзмо – Енвмо = E1/23– E1/2 1 = Еox-red (55)
По разности потенциалов Ered = E1/22 – E1/21 и Еox = E1/24 – E1/23 можно судить о смешении орбиталей лигандов и иона металла. Для комплексов 1-21 рассчитаны значения (Eox-red=0,560,92В, Еred=0,841,28В, Еox=0,280,78В), указывающие на энергетическую близость граничных орбиталей металла и лигандов. В связи с этим сделано предположение о возможности координации H2S и тиолов на металлоцентре комплексов, участвующих в редокс-активации серосодержащих реагентов.
- Применение комплексов металлов VIII группы с peдокс-активными лигандами в синтезе органических соединений серы
Электрохимическое поведение комплексов 1-15 свидетельствует о том, что они могут выступать в качестве одноэлектронных окислителей. К тому же, дианионная форма комплексов способна к регенерации в присутствии кислорода воздуха. Следовательно, комплексы 1-15 аналогично о-бензохинонам можно использовать для окисления H2S до катион-радикала.
В реакциях H2S с соединениями 1-15 наблюдается сдвиг потенциалов окисления комплексов в отрицательную область на 0,1-0,2 В в зависимости от его структуры, что обусловлено координацией H2S атомом металла. Далее происходит перенос электрона, приводящий к образованию катион-радикала H2S и восстановлению комплекса до моноанионной формы:
 (56)
(56)
Продукт фрагментации катион-радикала сероводорода (H+) стабилизирует образующийся моноанион комплекса и способствует генерированию координированной аминогруппы в лиганде. В ЭСП исчезают максимумы поглощения (750-890 нм), отвечающие переносу заряда в системе лиганд-лиганд. Для семихинондииминовых комплексов в присутствии H2S наблюдается возрастание по току второго пика окисления, что характеризуется каталитическим эффектом.
Значения констант скоростей взаимодействия сероводорода с комплексами 1-15 (k=1,04,5·103 с-1) свидетельствуют о наиболее высокой эффективности соединений 10-12 (k=2,84,5·103 с-1), которые восстанавливаются при низких потенциалах по сравнению с комплексами 1-9, 13, 14. Наименьшую эффективность продемонстрировали комплексы 7-9 (k=1,12,1·103 с-1) с донорными заместителями.
Несмотря на незначительную роль металла (ввиду постоянства степени окисления) его природа оказывает влияние на величину константы скорости реакции комплексов с H2S и изменяется в ряду: Ni(L)2>Pd(L)2>Pt(L)2. Скорость взаимодействия H2S с комплексами Pd выше, чем для платиновых и никелевых аналогов, которые чувствительны к избытку H2S. Разрушение комплексов в присутствии H2S фиксируется на ЭСП и по появлению на ЦВА пиков окисления свободных лигандов: о-фенилендиамина (0,46; 1,3 В), N-фенил-о-фенилендиамина (0,66; 1,46 В). Регенерация комплексов протекает по схеме:
 (57)
(57)
Образование нейтральных комплексов в результате электролиза соединений 16-21 свидетельствует о возможности их использования в качестве электромедиаторов для активации H2S до катион-радикала:
 (58)
(58)
Результаты исследования реакций H2S с комплексами 1-21 указывают на возможность их использования как окислителей H2S в синтезе органических соединений серы. Взаимодействие H2S с ароматическими соединениями Ic-IVc в присутствии комплексов 1-15 при 25 °С приводит к соответствующим тиофенолам и симметричным дисульфидам. Эффективным оказалось использование комплексов 1-12. Наибольший выход ароматических тиолов и дисульфидов независимо от строения субстрата наблюдается для комплексов платины 3, 6, 10. Природа растворителя существенно не влияет на состав и соотношение продуктов реакции. Реакционная способность комплексов 13-15 ниже и для них характерно большее время регенерации. Однако, ввиду наличия связи M-S комплексы с S,S-координированными лигандами обладают устойчивостью к избытку сероводорода.
Реакции ароматических соединений Ic, IIc с H2S были проведены в условиях электрохимической активации комплексов 16-21, необходимой для перевода их из моноанионного в нейтральное состояние. В результате получены соответствующие тиофенолы и дисульфиды, однако выход продуктов реакции снижается из-за адсорбции комплексов 16-21 на поверхности электрода. В роли окислителей H2S при взаимодействии с олефинами (октеном-1) были использованы комплексы 2, 3, при этом получены n-C8H17SH и (C8H17)2S (молярное соотношение 1,3 : 1).
В реакциях H2S с фураном рассмотрена эффективность 2, 3, 5, 6 и 13 лигандами. В результате реакции H2S с фураном в присутствии комплексов 3, 6 получены 2-тиофентиол (16,5 %) и бис(тиенил-)дисульфид (3 %). Взаимодействие H2S с фураном в присутствии комплекса 13 (3 ч) осуществляли при введении серы, однако, синтез тиофена (8,5 %) и 2-тиофентиола (1,2 %) сопровождался образованием, преимущественно, бис(тиенил)дисульфида и политиофенов. Такое направление маршрута реакции объясняется окислением 2-тиофентиола c последующей полимеризацией бис(тиенил)дисульфида.
В синтезе тиопиранов и тиопирилиевых солей на основе 1,5-дикетонов IIId-Vd, Xd, IXd и H2S были использованы комплексы 1-6, 13-15. Реакция S-циклизации в присутствии комплексов 2, 3, 6, 13, 15 при 25 °С приводит к соответствующим тиопиранам и солям (молярное соотношение 1 : 1). Применение комплексов 2, 5 для активации H2S в синтезе органических соединений серы приводит к их разрушению и образованию сульфидов металлов. Повысить устойчивость комплексов 2, 5 к сероводороду удается при разбавлении реагента инертным газом (1:3).
Таким образом, проведенные исследования показали эффективность использования комплексов переходных металлов VIII группы с редокс-активными лигандами в реакциях замещения в бензоле и его производных, тиолирования олефинов, S-циклизации 1,5-дикарбонильных соединений и S-гетероциклизации фурана c участием сероводорода.
- Использование органических медиаторов для активации сероводорода и алкантиолов в реакциях с олефинами и ароматическими углеводородами
В последнее время разработка новых синтетических методов, основанных на косвенной электрохимической активации молекул, актуальна ввиду возможности уменьшения энергозатрат и негативного влияния на окружающую среду. Имеются сведения об активации тиолов в присутствии Ph3P[7], однако информация о переводе H2S в катион-радикальное состояние с участием электромедиаторов отсутствует.
Для активации RSH (R=Н, Alk, H2Sn) до катион-радикалов рассмотрены органические электромедиаторы: замещенные ароматические амины 1а-9а и комплексы хрома, никеля и платины с редокс-активными лигандами 10а, 16, 18, 19, 21, которые ранее не применялись в качестве Меd:
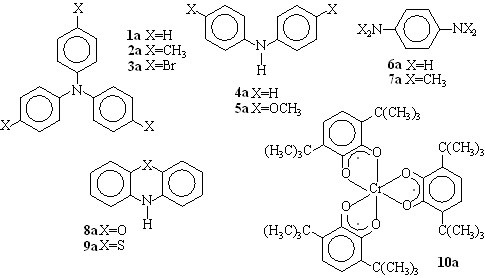
Исследуемые электромедиаторы 1a-10a, 16, 18, 19, 21 при сравнительно низких анодных потенциалах (0,7 - 1,0 В) образуют стабильные катион-радикалы (или радикалы) и способны к регенерации.
Таким образом, выбранные соединения 1a-10a, 16, 18, 19, 21 соответствуют требованиям, предъявляемым к потенциальным электромедиаторам[8]
. Для медиаторных систем (соединения 1a-10a, 16, 18, 19, 21 и RSH (R=Н, Alk, H2Sn) рассчитаны величины снижения энергетического барьера (E) электрохимической реакции (табл. 5).
Табл. 5
Разность потеницалов окисления сероводорода и медиаторов
| Med | 1a | 2a | 3a | 4a | 5a | 6a | 7a | 8a | 9a | 10a | 16 | 18 | 19 | 21 |
| E, В | 0,68 | 0,76 | 0,40 | 1,15 | 0,62 | 1,11 | 0,80 | 0,91 | 0,93 | 0,80 | 0,92 | 0,98 | 1,04 | 1,06 |
Примечание: Е=ER-EMed, где ER – потенциал окисления H2S, EMed – потенциал окисления электромедиатора
При активации H2S наибольшее снижение энергозатрат достигается при использовании медиатора 4a, а наименьшее – в случае соединения 3a. Аналогичная зависимость характерна и для реакций Меd c тиолами (сульфанами). Активация RSH (R = Н, Alk, H2Sn) электромедиаторами происходит по механизму ЕСЕ с обратимым переносом электрона на электрохимических стадиях (Iпк/Iпа1). Расчет значений констант скоростей взаимодействия H2S с электромедиаторами 1а-9а (k=0,031,19 c-1) позволил расположить производные ароматических аминов, катион-радикальные формы которых проявляют наибольшую активность, в следующий ряд:
(4-MeC6H4)3N > Ph3N > 1,4-Ph(NMe2)2> (4-BrC6H4)3N>(2,4-MeOC6H4)2NH
Значения константы скорости реакции для третичных и вторичных ароматических аминов 1а-4а, 7a соизмеримы. Природа заместителя в молекуле реагента оказывает существенное влияние на скорость его взаимодействия с медиаторами. Например, при использовании медиатора 2а наблюдается снижение значения константы скорости от k=1,19 с-1 до k=0,49 с-1 в ряду: H2S > C2H5SH > C4H9SH > C6H13SH > C8H17SH.
Было изучено поведение электромедиаторов 1a-9a в присутствии H2S и тиолов в условиях электрохимического и спектрофотометрического контроля. В реакции катион-радикала соединения 1a и гексантиола-1 при возрастании содержания RSH на ЦВА наблюдается уменьшение по току пика восстановления катион-радикала амина (при неизменном по току анодном пике), что свидетельствует о снижении обратимости редокс-процесса. При этом фиксируется образование катионов [RSH2]+ (-0,36 В) и [Ph3NH]+ (-1,85 В). Аналогичные результаты получены и для соединений 2a, 6a, что также указывает на протекание побочных процессов.
При исследовании медиатора 3a небольшое возрастание по току анодного пика объясняется низкой скоростью стадии восстановления Med вследствие депротонирования катиона [(4-BrC6H4)3NH]+. Для медиаторов 8a, 9a при увеличении концентрации реагента перенос электрона сопровождается каталитическими токами, возникающими из-за образования КПЗ:
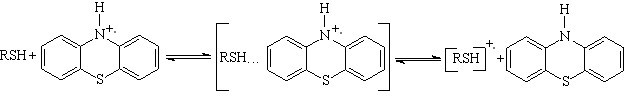 (59)
(59)
Электролиз смеси (RSH (R = H, Alk) + соединение 1a-9a) при потенциале окисления Med приводит в случае H2S (рис. 19) к сульфанам (выход по току 60%), а в реакции с тиолами – к дисульфидам (выход по току 87%).
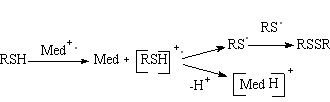
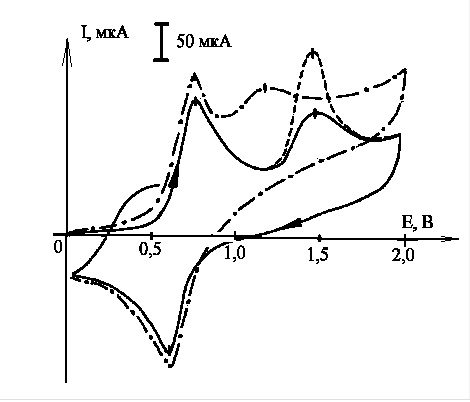 Рис. 19 – ЦВА соединения 8а _._._.; продукта реакции H2S с гексантиолом-1, полученного в присутствии 8а _____; идентификация С6Н13S2 добавкой стандарта (C=210-3 моль/л) - - - (Ag/AgCl, СН2Cl2, 0,1 М NBu4ClO4) Рис. 19 – ЦВА соединения 8а _._._.; продукта реакции H2S с гексантиолом-1, полученного в присутствии 8а _____; идентификация С6Н13S2 добавкой стандарта (C=210-3 моль/л) - - - (Ag/AgCl, СН2Cl2, 0,1 М NBu4ClO4) | Теплота образования сульфанов и дисульфидов равна -55,14 кДж/моль и -104,45 кДж/моль, соответственно. При добавлении RSH (R=Н, Alk) к электрогенерированным катион-радикальным солям Меd наблюдается гипcохромный сдвиг максимумa поглощения (max = 610 480 нм), отвечающего нейтральным соединениям 1a-9a. Общий принцип действия Med 1a-9a в реакциях с RSH (R = Н, Alk, H2Sn) в присутствии органических субстратов описывается схемой: |
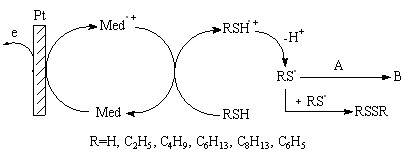
А – субстрат, В - продукт реакции
R = H, С2H5, n-C4H9, n-C6H13, n-C8H17 (60)
На примере соединения 7a схему активации H2S в присутствии электрогенерированной формы медиатора можно представить следующим образом:
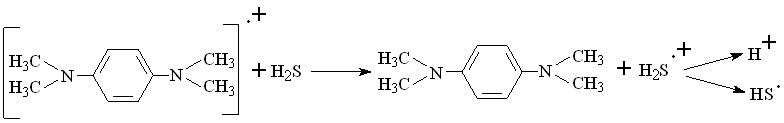 (61)
(61)
Реакция H2S с гексеном-1 в присутствии медиаторов 1a-9a (молярное соотношение Med : H2S = 1 : 8) приводит к получению н-гексантиола (выход по току 43 – 88%) и дигексилдисульфида (выход по току 12 – 43 %). В зависимости от природы медиаторов в реакции H2S с октеном-1 выход по току варьируется: н-октантиол-1 (45 – 91 %) и диоктилдисульфид (8 – 42 %). Электролиз смеси (n-С6H13SH + гексен-1) в присутствии активированных на аноде медиаторов 1a-9a (молярное соотношение Med : RSH = 1 : 20) приводит к дигексилсульфиду и дигексилдисульфиду (выход по току 18 – 90,6 %). В реакции исследуемых тиолов с октеном-1 в присутствии медиаторов 1a-9a образуются соответствующие сульфиды (выход по току 80 %).
Превращения олефинов в присутствии медиаторов 1a-9a и RSH (R=Н, Alk) можно описать схемой:
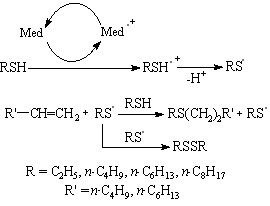 (62)
(62)
Взаимодействие H2S с бензолом в условиях электрохимического окисления медиаторов 2а, 3а, 5а, 7а приводит к образованию тиофенола (выход по току 33 – 36 %). При использовании катион-радикалов соединений 8a, 9a для окисления H2S в реакции с бензойной кислотой получены тиосалициловая кислота и симметричный ароматический дисульфид. Взаимодействие H2S с толуолом в присутствии медиаторов 8a, 9a приводит к смеси изомерных тиокрезолов, бензилтиола и дисульфида Ar-S-S-Ar:
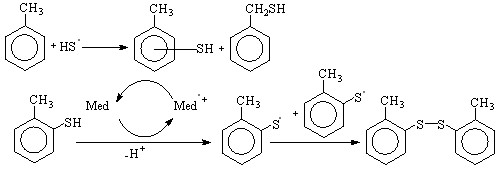 (63)
(63)
С целью получения ассиметричных дисульфидов и сульфидов исследованы реакции тиолов (C2H5SH, n-C4H9SH, n-C6H13SH) с ароматическими соединениями в присутствии медиаторов 2a, 3a, 5a, 7a (молярное соотношение Med : H2S = 1 : 8):

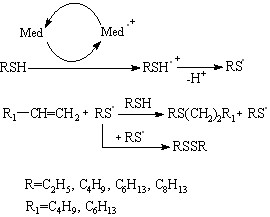 (64)
(64)
Образование тиокрезола подтверждено данными ИК-спектроскопии: в спектрах фиксируются валентные (=2580 см-1), деформационные (=850 см-1) колебания SH-связей и валентные колебания (=610 см-1) С-S связи. Методом хроматомасс-спектрометрии зафиксирован перенос атома водорода от замещенной группы –СOOH к группе –SH с отщеплением H2S и образованием стабильного иона m/e – 136.
Реакция нитробензола с тиильным радикалом, генерированным в присутствии катион-радикалов медиаторов 1a-9a, приводит к образованию тиозамещенных продуктов O2N-C6H5SH (1,90 В) и O2NC6H5-S-S-C6H5NO2 (1,68 В). При избытке H2S, как и в условиях прямого способа его электрохимической активации в реакции с нитробензолом, протекает конкурирующая стадия реакции – образование анилина за счет перенапряжения на катоде:
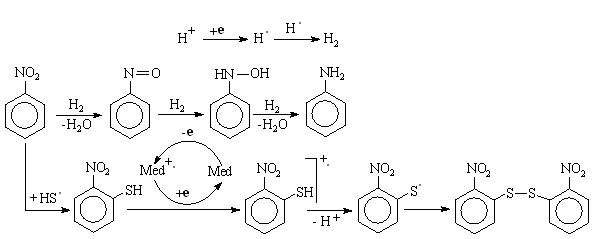 (65)
(65)
Из серии комплексов с редокс-активными лигандами наиболее эффективными оказались электромедиаторы 10a, 16, 19, в которых степень окисления никеля и хрома не изменяется в ходе редокс-процесса. Комплексы платины 18, 21 при избытке H2S разрушаются с образованием сульфидов металлов. Эффективность комплексов 16, 19 как электромедиаторов по сравнению с ароматическими аминами 1a-9a выше за счет предварительной координации молекул RSH (R = Н, Alk) на металлоцентре. В случае комплекса хрома 10a вследствие стерических затруднений координация H2S невозможна[9]
. При окислении комплексов никеля 16, 19 с 1,2-бензо-дитиолатными лигандами активной формой по отношению к сероводороду является бирадикальная система, а для трис-о-семихинолятного комплекса хрома 10a - о-бензохиноновая форма.
Взаимодействие H2S и тиолов с алифатическими (гексеном-1, октеном-1) и ароматическими (бензолом, толуолом, бензойной кислотой) соединениями в присутствии комплексов 10a, 16, 19 приводит к образованию соответствующих тиолов и сульфидов (выход по току 62 – 75 %). Таким образом, обнаружен различный механизм активации H2S электромедиаторами 10a, 16, 19.
В случае соединений 1а, 4а, 6a, 9а реакции осложняются выводом медиаторов из цикла электрогенерирования в связи с их участием в параллельных превращениях. Так, при взаимодействии электромедиатора 1а с серосодержащими реагентами RSH (R = Н, Alk) образуется N,N,N`,N`- тетрафенилбензидин (0,75 В):
![]()
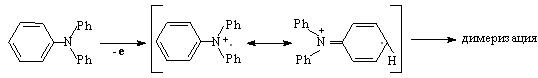 (66)
(66)
Реакция сероводорода и тиолов с соединением 4а сопровождается димеризацией аминильных радикалов с образованием тетрафенилгидразина. Логично было ожидать, что при взаимодействии RSH (R = Н, Alk) с медиатором 4а будет происходить тиолирование незамещенного ароматического кольца Med. Однако, анализ продуктов реакции и квантово-химический расчет (Нобр = 154 кДж/моль) подтверждают отсутствие тиопроизводных соединения 4а. Реакция соединения 9a с H2S приводит к сульфанам и продукту электрополимеризации фенотиазина с 2,2’-замещенными фениленовыми фрагментами.
Для электромедиатора 6a характерны низкие значения потенциала окисления (0,50 В) и коэффициента обратимости редокс-процесса. В отличие от соединений 1a-5a, 8a, 9a п-фенилендиамин окисляется в две одноэлектронные стадии до дикатиона, который реагирует с исходной молекулой Med с образованием катион-радикала:
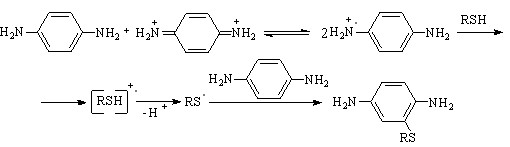
R = H, С2H5, n-C4H9, n-C6H13, n-C8H17 (67)
В результате взаимодействия соединения 6a с RSH (R = Н, Alk) получен продукт G радикального замещения в ароматическом кольце п-фенилендиамина. Электрохимические превращения соединения G в присутствии H2S приводят к диаминопроизводному фенотиазина H. На ЭСП фиксируется батохромный сдвиг максимума поглощения (maх = 260320 нм) по сравнению с мах для незамещенного фенотиазина 9a. Предполагаемый механизм образования 10H-фенотиазин-3,7-диамина H описывается схемой:
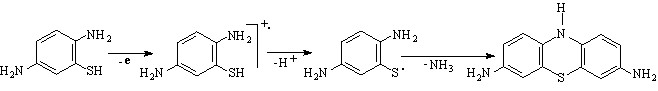
(68)
Квантово-химический расчет (GAMESS, метод PM3) показал, что образование 10H-фенотиазин-3,7-диамина по реакции Н2S с электромедиатором 7a достаточно выгодно (Н = -30,5 кДж/моль). Методом хроматомасс-спектрометрии (ЭУ, 70 эВ) фиксируются ионы (Iотн,%): 229 [M]+ (100), 197 [M–S]+ (50), 136 [M–Ph–NH2]+ (8), 121 [M–NH–Ph–NH2]+ (5). Осколочные фрагменты характеризуют распад 10H-фенотиазин-3,7-диамина с элиминированием S, CNH и диссоциацией связи Ar-N.
Проведенные исследования позволили установить взаимосвязь природы радикала и скорости реакции с электромедиаторами 1a-14a в ряду серосодержащих реагентов RSH (R = Н, Alk). В присутствии медиаторов 2a, 3a, 5a, 8a из Н2S и серы образуются сульфаны (выход по току 90 %):
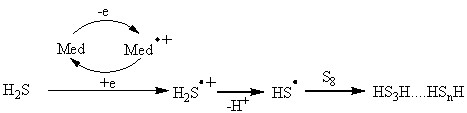 (69)
(69)
В случае тиолов RSH (R = Н, C2H5, C4H9, C6H13, C8H17, C6H5) использование медиаторов приводит к симметричным дисульфидам. Методом электронной растровой микроскопии установлено, что при деструкции сульфанов образуется молекулярная сера, представляющая собой наноразмерные ассоциаты (100 - 200 нм).
Электролиз трехкомпонентной системы «HSnH – медиатор 2a – циклогексен (циклогексадиен)» приводит к сульфанам (1,4 - 1,6 В) циклического строения. Квантово-химический расчет показал, что образование циклических форм энергетически выгоднее (в случае циклогексена на 447 кДж/моль) по сравнению со значением H для линейных сульфанов. В результате электролиза H2Sn с циклогексеном в присутствии медиатора 2a выделен продукт реакции: в ИК-спектре наблюдаются валентные колебания связей – 532 см-1 (S-S), 685 см-1 (С-S) и 2900 см-1 (С-H в циклоалкановом кольце). Рентгенофлуоресцентный анализ на наличие серы в смеси продуктов реакции подтвердил образование тетратиепино[1,2-]циклогексана (дитетратиепино[1,2,4,5-]циклогексана) из циклогексена (циклогексадиена):
 (70)
(70)
Таким образом, впервые предложен эффективный способ непрямого окисления реагентов RSH (R = H, Alk, Н2Sn) в присутствии электрогенерированных катион-радикалов медиаторов. Это позволило разработать новый одностадийный метод синтеза органических соединений серы с участием сероводорода, тиолов и сульфанов в энергетически выгодных условиях. Перспективность применения металлокомплексов никеля и хрома с редокс-активными лигандами в качестве электромедиаторов обусловлена усилением их функции за счет координирования молекул металлоцентром. Продемонстрирована возможность реакции аннелирования тетратиенпинового цикла к шестичленным насыщенным и ненасыщенным циклическим углеводородам на основе электрогенерированных органическими медиаторами сульфанов.
- Синтез органических соединений серы с участием сероводорода в гетерогенных условиях
Проведенные исследования показали перспективность использования 3,5-ди-трет-бутил-о-бензохинона Q1 и комплексов металлов с редокс-активными лигандами 1, 3, 4-6, 13, 15 в качестве окислителей сероводорода. Регенерацию окислителей кислородом воздуха удобно реализовать в условиях циркуляции потоков в кварцевом реакторе, что позволяет повысить выход органических соединений серы до 60 – 70 % ввиду увеличения количества циклов «реакция-регенерация».
Для синтеза органических соединений серы в гетерогенных условиях окислители наносились на поверхность цеолита (Na2OAl2O3xSiO2), оксид алюминия (-Al2O3) или силикагеля (SiO2nH2O) путем пропитки. Выбранные носители без модифицирующих добавок обладают активностью в синтезе алифатических, ароматических и гетероциклических серосодержащих соединений при 250500 °С.
При проведении реакции H2S с гексеном-1 на цеолите, модифицированном Q1, при комнатной температуре образуются гексантиол-1 и дигексилсульфид (молярное соотношение 0,5 : 1). Низкий выход гексантиола-1 (10 %) объясняется достаточно узкими "входными окнами" (0,4 нм) носителя и адсорбцией в них продуктов реакции. Аналогичная реакция на поверхности (-Al2O3+Q1) приводит к увеличению выхода гексантиола-1 (молярное соотношение RSH : RSR – 3,4 : 1). Негативным фактором использования -Аl2О3 (50 - 85 нм) является десорбция Q1 с поверхности носителя. По данным квантово-химических расчетов адсорбционный комплекс (-Al2O3+Q1) образуется за счет слабой донорно-акцепторной связи Al - O: rAl-O = 0,25 нм. В ходе реакции поверхность -Аl2О3 дезактивировалась молекулярной серой.
В качестве конкуретного носителя для модификации окислителем Q1 использовали силикагель ШКСМ (8 - 10 нм). Длина связи в адсорбционном комплексе (Q1+SiO2nH2O) между атомами кислорода о-бензохинона и водорода SiOH-группы равна 0,19 нм, что характерно для сильной водородной связи.
Реакцию H2S с гексеном-1 на поверхности (SiO2nH2O+Q1) проводили при 25, 50, 75 и 100 С. Степень превращения гексена-1 в серосодержащие продукты реакции (гексантиол-1, дигексилдисульфид, дигексилсульфид) увеличивается при повышении температуры. Однако селективность образования тиола снижается ввиду возрастания скорости реакций присоединения гексантиола-1 к субстрату и окисления тиола до дисульфида. Наиболее целесообразным оказалось проведение синтеза серосодержащих соединений на основе олефинов и H2S при 50 °С с использованием 3,5-ди-трет.-бутил-о-бензохинона, нанесенного на силикагель.
 Рис. 20 - Зависимость выхода соединений n-С6Н13SH, (C6H13)2S, (C6H13)2S2, полученных по реакции H2S с толуолом в присутствии Q1 от температуры. Рис. 20 - Зависимость выхода соединений n-С6Н13SH, (C6H13)2S, (C6H13)2S2, полученных по реакции H2S с толуолом в присутствии Q1 от температуры. | Реакцию H2S c бензолом (толуолом) проводили на -Аl2О3, модифицированном Q1, (рис. 20) и комплексами 1, 3, 4-6, 13, 15 при 30, 45, 60, 75 и 90 0С. Повышение температуры способствует увеличению выхода при неизменном соотношении продуктов. Рециклизацию фурана в присутствии H2S проводили на -Al2O3, модифицированном Q1 или комплексом 16 (рис. 19). Содержание Q1 на поверхности -Al2O3 составляло 1 и 5% мас. при 25, 50 и 75С. При использовании Q1 (1%) выход |
тиофена увеличивается в зависимости от температуры (1 : 2,8 : 3,7). Кинетические зависимости для реакции H2S с фураном на -Al2O3, промотированном окислителем Q1, демонстрируют возрастание константы скорости при увеличении температуры от 25 до 75С (рис. 20). Рециклизацию ТГФ в присутствии H2S проводили на -Al2O3, модифицированном Q1 или комплексом 16.
 Рис. 19 – ЦВА смеси продуктов реакции H2S с фураном в присутствии Q1 (5%) (=2 ч, t=75С, Ag/AgCl, СН2Cl2, 0,1 М NBu4ClO4) Рис. 19 – ЦВА смеси продуктов реакции H2S с фураном в присутствии Q1 (5%) (=2 ч, t=75С, Ag/AgCl, СН2Cl2, 0,1 М NBu4ClO4) | 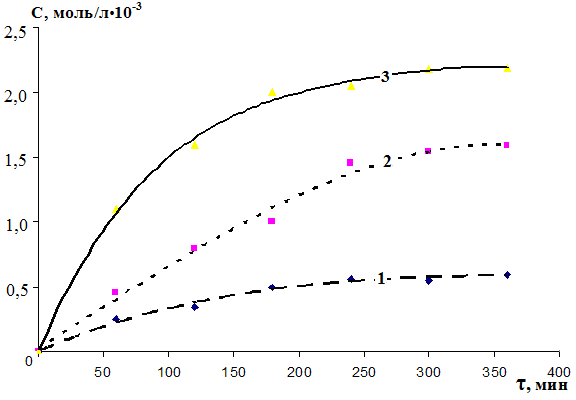 Рис. 20 – Кинетические кривые образования тиофена в реакции сероводорода с фураном при 25 (1), 50 (2) и 75С (3) Рис. 20 – Кинетические кривые образования тиофена в реакции сероводорода с фураном при 25 (1), 50 (2) и 75С (3) |
При анализе продуктов реакции сероводорода с ТГФ методом масс-спектрометрии зарегистрированы сигналы молекулярного иона m/z 88 [М]+ и фрагментных ионов, подтверждающих рециклизацию ТГФ в тиофан. На ИК-спектре наблюдались валентные колебания 676, 687 см-1 (C–S) и 2865, 2956 см-1 (С–Н).
Таким образом, активация H2S до катион-радикала при модификации одноэлектронными окислителями носителей Na2OAl2O3xSiO2, -Al2O3, SiO2nH2O, позволяет значительно снизить температуру синтеза с участием сероводорода по сравнению с традиционными условиями и увеличить выход органических соединений серы.
- практические аспекты
Электрохимическая и химическая активация H2S при взаимодействии с фураном (пирролом) в органических средах приводит к тиофену и его производным, а далее – продуктам полимеризации соединений тиофенового ряда. В связи с этим были рассмотрены пути интенсификации процесса получения политиофенов, широко применяемых в электронной промышленности.
Электрополимеризация тиофена, образующегося в реакции H2S с фураном (пирролом), протекает в приэлектродном слое вследствие фрагментации катиона-радикала H2S с отрывом протона:
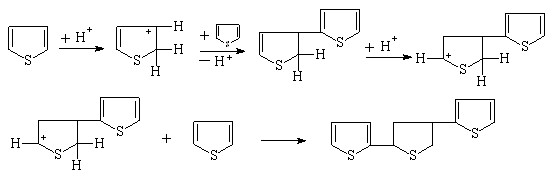 (71)
(71)
Окисление бис(2-тиенил)дисульфида, полученного при взаимодействии H2S с фураном (пирролом) также способствует росту полимерной цепи:
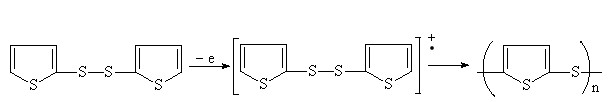 (72)
(72)
Выход политиофенов снижается при проведении электролиза в инертной атмосфере и замене растворителя (СН3СNСH2Cl2). Значительно ускоряется полимеризация тиофена в CH3CN (5% H2O) и при увеличении площади поверхности анода (выход политиофенов 14 %).
Применение комбинированного электрохимического и химического воздействия на H2S в реакции с фураном приводит к увеличению выхода политиофенов. Окислитель Q1 в ходе электролиза смеси (H2S+фуран) способствует повышению конверсии тиофена, 2-тиофентиола и бис(2-тиенил)дисульфида в политиофены до 100% и увеличению молекулярной массы полимера. На поверхности анода образуется полимерная пленка, включающая перхлорат-анионы (данные ИК-спектроскопии), что обуславливает электропроводимость полученных политиофенов за счет движения носителей заряда по системе сопряженных связей:
 (73)
(73)
Таким образом, механизм электрополимеризации (Е(СЕ)n) тиофена и его производных в рассматриваемых условиях включает чередование электрохимических и химических стадий с участием тиофена и активированных H2S, 2-тиофентиола и бис(2-тиенил)дисульфида.
Рециклизация O-, N-гетероцикличексих соединений с участием H2S в присутствии окислителей Q1-Q5 также приводит к продуктам полимеризации тиофена и его производных. В ходе реакции H2S с пирролом при активации реагента о-бензохиноном Q1 наряду с политиофеном (3 % мас.) получен полипиррол (10 % мас.).
В последнее время для интенсификации процесса полимеризации органических соединений широко используется молекулярная сера. При введении серы (5 % мас.) удается значительно повысить выход политиофена (24%) в реакции H2S с фураном в присутствии Q1 при 25°С. Полученный полимер окисляется при Епа=1,58 В (tпл=55°С, Мr=57200 г/моль). С увеличением содержания серы (10 % мас.) в реакционной смеси выход и вязкость политиофена возрастают. ИК-спектр полимера (колебания S–S- (545, 568 см-1) и C–S-связей (613 см-1)) и определение содержания серы в полимере методом РФА позволии предложить структуру композита как тиофеновые циклы, связанные дисульфидными мостиками.
Очистка углеводородного сырья и газовой серы, получаемой по методу Клауса, от агрессивных и токсичных компонентов актуальна в связи с негативным влиянием их на качество продукции. На основании проведенных исследований путей активации серосодержащих соединений RSH (R=H, Alk, Н2Sn) предложены новый эффективный способ перевода тиолов в менее токсичные дисульфиды, а также сероводорода и сульфанов в молекулярную серу в присутствии замещенных о-бензохинонов при комнатной температуре. Использование одностадийного маршрута реакции с применением одноэлектронных окислителей позволяет развивать новое направление в химии и технологии органических соединений серы.
ВЫВОДЫ
- Создано новое научное направление в химии органических соединений серы – окислительная активация сероводорода, алкантиолов и сульфанов в реакциях с органическими соединениями. Развита концепция повышения реакционной способности RSH (R = H, Alk, HSn) посредством одноэлектронного окисления в неводных средах и предложены основные пути ее использования в органическом синтезе. Установлено, что продукты окислительной активации RSH (R = H, Alk, HSn) – нестабильные катион-радикалы – обладают свойствами сильных кислот и способны к фрагментации с образованием протонов, (алкил-)тиильных и полисульфидных радикалов.
- Разработана общая стратегия синтеза органических соединений серы в условиях окислительной активации серосодержащих реагентов RSH (R = H, Alk, HSn) и определены ключевые интермедиаты, генерируемые прямым и косвенным электрохимическими способами, в присутствии одноэлектронных окислителей (о-бензохинонов, комплексов металлов VIII группы с редокс-активными лигандами) или при использовании комбинированных систем (анод – химический реагент).
- Впервые осуществлено тиолирование ароматических и непредельных соединений в присутствии электрохимически активированного сероводорода по радикальному механизму при 25 °С с полученем тиопроизводных бензола, толуола, фенолов, нитробензола, бензальдегида, анилина и бензойной кислоты, а также (гексан-, гептан-)октантиолов-1 и соответствующих сульфидов и дисульфидов.
- Предложены новые методы получения (алкил-)арилзамещенных тиопиранов и солей тиопирилия по реакции S-циклизации 1,5-дикарбонильных соединений в присутствии анодногенерированной окисленной формы H2S и установлено влияние потенциала окисления субстрата на спектр продуктов электросинтеза. Изучены превращения O-, S-, Se-содержащих 1,5-дикарбонильных соединений в реакциях с катион-радикалом сероводорода.
- Разработаны методы прямого электросинтеза соединений тиофенового ряда и политиофенов на основе пятичленных O-, N-содержащих гетероциклов и сероводорода в условиях электрохимической активации субстрата и реагента, а также изучено влияние природы субстрата и растворителя на выход продуктов рециклизации фурана, 2,5-диметилфурана и пиррола в серосодержащие аналоги.
- Показано, что реакции пиридина и его конденсированных аналогов с электрохимически активированной формой сероводорода приводят к соответствующим катионам и изомерным 1,2-, 1,4-тиодигидропроизводным. Рассмотрена рециклизация оксатиинов с конденсированными бензольными фрагментами в присутствии катион-радикала сероводорода и исследованы побочные превращения субстратов при проведении электросинтеза.
- Предложен простой и удобный метод синтеза производных серы алифатического, ароматического и гетероциклического рядов при использовании пространственно-затрудненых о-бензохинонов как одноэлектронных окислителей сероводорода, алкантиолов и сульфанов. Выявлен критерий оценки эффективности окислителей и показано, что использование системы «сероводород – сера – о-бензохинон» способствует повышению выхода органических соединений серы и конверсии субстратов в продукты реакции.
- Рассмотрены моноядерные комплексы металлов VIII группы (никеля, палладия и платины) с редокс-активными N,N-, N,S- и S,S-координированными лигандами в качестве активаторов сероводорода. Выявлено участие редокс-форм лигандов в его одноэлектронном окислении, установлено влияние природы металлоцентра на устойчивость комплексов в присутствии H2S и предложен на этой основе метод синтеза (алкил-)арилзамещенных тиопиранов, тиопирилиевых солей, тиофенола, изомерных тиокрезолов, тиофена, гексантиола-1.
- Впервые рекомендовано использование в качестве электромедиаторов комплексов никеля и хрома с семихинондитионовыми лигандами и показана возможность применения в этих целях замещенных ароматических аминов, снижающих потенциал электролиза в реакциях сероводорода, тиолов и сульфанов с ароматическими и ненасыщенными углеводородами.
- Предложены комбинированные системы «анод – о-бензохинон», «анод – основание Льюиса» для снижения энергетического барьера реакций сероводорода с органическими соединениями, позволяющие проводить электросинтез их серопроизводных при низких значениях анодных потенциалов.
- Впервые осуществлен синтез органических соединений серы с участием сероводорода при 25 – 100 °С благодаря модификации поверхности -Al2O3, Na2OAl2O3xSiO2 и SiO2nH2O о-бензохинонами и комплексами металлов VIII группы с редокс-активными лигандами. Наибольший выход продуктов реакции достигается при использовании SiO2nH2O, содержании окислителя – 5 % мас., 75 °С.
- Совокупность полученных данных положена в основу разработки способов демеркаптанизации углеводородного сырья и деструкции сульфанов, содержащихся в газовой сере, а также способа получения серы на основе сероводорода с использованием различного типа активирующих агентов.
Основное содержание диссертации изложено в следующих работах:
Монография и статьи по перечню ВАК:
- Берберова Н.Т., Шинкарь Е.В., Смолянинов И.В., Охлобыстин А.О.. Вовлечение сероводорода, тиолов и полисульфанов в синтез органических соединений серы. Монография, Ростов-на-Дону: ЮНЦ РАН, 2009. – 256 c.
- Берберова Н.Т., Летичевская Н.Н., Фоменко А.И., Милаева Е.Р., Шинкарь Е.В., Прокофьев А.И. Катион-радикал сероводорода и реакции циклизации 1,5-дикетонов с его участием // Электрохимия. – 2000. – Т. 36. – № 2. – С. 203 -209.
- Берберова Н.Т., Шинкарь Е.В. Катион-радикал сероводорода и органические реакции с его участием // Известия РАН, Серия химическая. 2000. – № 7. – С.1182-1188.
- Талисманова М.О., Ерёменко И.Л., Сидоров А.А., Фомина И.Г., Шинкарь Е.В., Охлобыстин А.О., Голованёва И.Ф., Александров Г.Г., Моисеев И.И.. Синтез и строение нового биядерного комплекса палладия, не содержащего мостиковых лигандов // Известия Академии наук. Серия химическая. – 2003. – Т. 52. – №12. – С. 2701-2706.
- Берберова Н.Т., Клименко С.К., Шинкарь Е.В., Акимова О.Н., Шарафутдинов Д.Р., Пащенко К.П. Электрохимическое получение и изучение редокс-превращений 2,4-дифенил-6Н-циклопента[b]тиопирана и перхлората 2,4- дифенилциклопента[b]тиопирилия // Электрохимия. – 2003. – Т. 39. – №12. – С. 1437-1443.
- Берберова Н.Т., Шинкарь Е.В., Фоменко А.И., Осипова В.П., Маняшин А.О., Зиньков Ф.Е. Роль одноэлектронных медиаторов в превращении сероводорода и сульфанов в элементарную серу // Известия ВУЗов. Химия и химическая технология, Иваново: ИГХТУ. – 2003. – Т. 46. – Вып. 6. – С. 74-78.
- Берберова Н.Т., Гиренко Е.Е., Шинкарь Е.В., Пащенко К.П., Разуваева А.В. Функционализация алкенов сероводородом в присутствии различных окислителей // Вестник ВУЗов. Химия и химическая технология, Иваново: ИГХТУ. – 2004. – Т. 47. – Вып. 8. – С. 10-13.
- Великородов А.В., Мухин А.А., Шинкарь Е.В. Взаимодействие N1-бензилоксикарбонил, N4-бензоил-п-бензохинондиимина с этилацетоацетатом // Известия ВУЗов. Химия и химическая технология, Иваново: ИГХТУ. – 2004. – Т.46. – Вып. 2. – С. 50-53.
9. Охлобыстин А.О., Берберова Н.Т., Шинкарь Е.В., Осипова В.П. Новый способ получения 2,6-ди-трет-бутил-4-гидроксифенилмеркаптана // Вестник Астраханского государственного технического университета.– 2005. – №6. – С. 40-44.
10. Берберова Н.Т., Шинкарь Е.В., Разуваева А.В. Исследование взаимодействия катион-радикалов аминов с тиолами и сероводородом // Вестник АГТУ, Астрахань. 2005. – №6(29). С. 29-34.
11. Хохлов В.А., Шинкарь Е.В., Берберова Н.Т., Маняшин А.О. Окислительное инициирование реакции получения тиофена на основе сероводорода и фурана // Вестник АГТУ. – Астрахань: АГТУ. – 2005. – Вып.6(29). – С. 75-81.
- Берберова Н.Т., Смолянинов И.В., Охлобыстин А.О., Летичевская Н.Н., Шинкарь Е.В. Структурные особенности и электрохимические характеристики комплексов переходных металлов (Pt, Pd, Ni, Co) с «небезучастными» лигандами // Российский химический журнал (Журнал Российского химического общества им. Д.И. Менделеева). - 2005. т. XLIX. - №5. - С. 67-74.
- Охлобыстин А.О., Берберова Н.Т., Шинкарь Е.В.. Новый каталитический способ получения тиопиранов и тиопирилиевых солей из 1,5-дикетонов и сероводорода // Вестник Астраханского государственного технического университета. – 2006. – №6. – С. 30-34.
- Аксенов А.В., Берберова Н.Т., Шинкарь Е.В., Охлобыстин А.О.Электрохимические превращения 2,2’-, 2,3’- и 3,3’-бихинолинов и их реакций с сероводородом // Вестник АГТУ, Астрахань. – 2006. – №6 (35). – С.15-22.
- Федотова О.В., Шинкарь Е.В., Пащенко К.П., Арефьев Я.Б. Электрохимические превращения халькогенбисцикланонов в органических средах // Вестник АГТУ, Астрахань. – 2007. – №6 (39). – С.145-149.
- Берберова Н.Т., Разуваева А.В., Шинкарь Е.В.Влияние структуры медиатора на эффективность электрохимических реакций с участием сероводорода и алифатических меркаптанов // Вестник АГТУ, Астрахань. – 2007. – №6 (39). – С.140-144.
- Берберова Н.Т., Шинкарь Е.В., Ахмедова Ю.И. Исследование действия окислителей на процесс разложения полисульфанов в газовой сере // Вестник АГТУ, Астрахань. – 2007. – №6(41). – С.8-13.
- Берберова Н.Т., Шинкарь Е.В., Фоменко А.И., Cмолянинов И.В., Ахмедова Ю.И. Окислительная деструкция полисульфанов // Известия вузов. Северо-Кавказский регион. Естественные науки (Спец. выпуск). – 2008. – С.39-42.
- Хохлов В.А., Берберова Н.Т., Шинкарь Е.В., Маняшин А.О., Алехина Ю.Ю. Вовлечение катион-радикала сероводорода в реакции с пятичленными гетероциклами // Известия ВУЗов. Химия и химическая технология, Иваново: ИГХТУ. – 2008. – Т.51. – Вып.6. – С. 45-48.
- Шинкарь Е.В., Хохлов В.А., Берберова Н.Т. К вопросу об утилизации сероводорода газоконденсатных месторождений в сернистые гетероциклы // Защита окружающей среды в нефтегазовом комплексе. – 2008. – №5. – С. 83-87.
- Охлобыстин А.О., Охлобыстина А.В., Шинкарь Е.В., Берберова Н.Т., Еременко И.Л. Электромедиаторы в синтезе органических соединений серы на основе сероводорода и тиолов // Доклады Академии наук. – 2010. – Т.435. – №3. – С.1-5.
- Шинкарь Е.В., Берберова Н.Т., Федотова О.В., Пащенко К.П., Арефьев Я.Б. Электрохимические реакции халькогенбисцикланонов и продуктов их S-, N-, O-гетероциклизации // Электрохимия. – 2011. – Т. 17. – № 10. – С. 1-8.
- Охлобыстина А.В., Шинкарь Е.В., Охлобыстин А.О., Берберова Н.Т., Абдулаева В.Ф. Катион-радикалы п-фенилендиаминов в синтезе органических соединений серы // Известия ВУЗов. Химия и химическая технология, Иваново: ИГХТУ. – 2011. – Т. 54. – №10. – С. 81-84.
- Шинкарь Е.В., Охлобыстина А.В., Колдаева Ю.Ю., Васильева Е.А., Петрова Н.В., Смолянинов И.В., Берберова Н.Т. Методы «зеленой химии» в утилизации серосодержащих компонентов углеводородного сырья // Защита окружающей среды в нефтегазовом комплексе. – 2011. – №10. – С. 67-71.
Патенты РФ:
- Пат. 207559 РФ, МПК 7G01N 27/48. Способ количественного определения меркаптанов в неводных средах/ Берберова Н.Т., Белинский Б.И., Тараканов Г.В., Шинкарь Е.В., Маняшин А.О., Гиренко Е.Е. – № 202113128, приоритет 18.05.2002, опубл. 27.06.2003, бюл. № 18.
- Пат. 2378644 РФ, МПК G01N 27/48. Способ количественного определения полисульфанов в газовой фазе / Берберова Н.Т., Шинкарь Е.В., Маняшин А.О., Леонова Ю.И. – №2006142262, приоритет 29.11.2006, опубл. 10.01.2010, бюл. №1.
- Пат. 2340609 РФ, МПК С07D 333/10, С07D 333/34. Способ получения тиофена и 2-тиофентиола в органических растворителях / Берберова Н.Т., Хохлов В.А., Шинкарь Е.В., Алехина Ю.Ю. –№2007134875, приоритет 19.09.2007, опубл. 10.12.2008, бюл. № 34.
- Пат. 2427608 РФ, МПК С10G 29/00. Способ демеркаптанизации углеводородного сырья / Берберова Н.Т., Шинкарь Е.В., Полякова Н.В. – №2009130801, приоритет 10.08.2009, опубл. 27.08.2011, бюл. № 24.
- Заявка на изобретение №2009130800, МПК CO1B17/16. Способ деструкции полисульфанов в товарной сере / Берберова Н.Т., Шинкарь Е.В., Смолянинов И.В., Охлобыстин А.О., опубл. заявки 20.02.2011. Получено положительное решение о выдаче патента.
Статьи:
- Берберова Н.Т., Фоменко А.И., Шинкарь Е.В., Маняшин А.О. Использование окисленной формы сероводорода в синтезе ароматических меркаптанов // Наука производству. – 2001. – №6 (44). – С. 54-55.
- Берберова Н.Т., Шинкарь Е.В., Маняшин А.О. Новые пути синтеза ароматических меркаптанов с участием сероводорода // Сборник статей IV Всероссийской научно-технической конференции "Новые химические технологии: производство и применение". Пенза. – 2002. – С.138-141.
- Шинкарь Е.В., Гиренко Е.Е., Берберова Н.Т. Синтез и кислотные свойства алкантиолов // Сборник статей IV Всероссийской научно-технической конференции "Новые химические технологии: производство и применение". Пенза. – 2002. – С. 91-92.
- Берберова Н.Т., Шинкарь Е.В., Охлобыстин А.О., Маняшин А.О. Новые подходы к использованию сероводорода газовых месторождений // Успехи современного естествознания. – 2003. – №4. – С. 76.
- Берберова Н.Т., Тараканов Г.В., Шинкарь Е.В. Сероводород Астраханского газоконденсатного месторождения как ценное сырье в синтезе сероорганических соединений
// Сборник научных трудов АстраханьНИПИгаза «Разведка и освоение нефтяных и газоконденсатных месторождений», Астрахань. -2003. – С. 131-134.
- Берберова Н.Т., Шинкарь Е.В., Охлобыстин А.О., Ерёменко И.Л., Сидоров А.А., Фомина И.Г. Каталитические свойства комплексов платины и палладия с семихинондиаминовыми лигандами в синтезе тиокрезолов // Вестник Астраханского государственного технического университета. – 2004. - №4. - С.24-28.
- Хохлов В.А., Шинкарь Е.В., Берберова Н.Т. Олиго- и полимеризация тиофена и его меркаптопроизводных в присутствии активированного сероводорода // Вестник АГТУ, Астрахань: АГТУ. – 2008. – Вып. 6 (47). – С. 23-30.
- Разуваева А.В., Шинкарь Е.В., Берберова Н.Т. Электроокисление ароматических аминов в синтезе органических соединений серы // Вестник АГТУ, Астрахань. 2008. 6(47). С. 31-36.
- Берберова Н.Т., Шинкарь Е.В., Колдаева Ю.Ю. Электрохимическое инициирование реакций пиридина и его бензаннелированных аналогов с сероводородом // Вестник АГТУ, Астрахань. 2008. 6(47). С. 31-36.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (гранты №№ 00-03-32911, 03-03-32256-а, 06-03-32442-а, 07-03-12101-офи 09-03-12122-офи-М, 09-03-00677-а). Настоящая работа является частью научных исследований, проводимых на кафедре органической, биологической и физколлоидной химии АГТУ. Разработка «Новый способ определения и удаления серосодержащих примесей в углеводородной среде и газовой сере» награждена золотой медалью на V Московском Международном салоне инноваций и инвестиций (2005 г.), разработка «Новый способ получения тиофена на основе отходов переработки растительного сырья» награждена серебряной медалью на XII Международном салоне промышленной собственности «Архимед» (2009 г.) и золотой медалью на Х Московском Международном салоне инноваций и инвестиций (2010 г.).
Благодарности.
За содействие в обсуждении результатов:
Научному консультанту по работе — д.х.н., проф. Берберовой Н.Т.
За сотрудничество и предоставление объектов исследования:
Д.х.н., проф. Федотовой О.В. и сотруд. (СГУ им. Н.Г. Чернышевского, г. Саратов)
За помощь в проведении хроматомасс-спектрометрического анализа:
Д.х.н., проф. Аксенову А.В. и сотруд. (СГУ, г. Ставрополь)
За помощь в проведении экспериментальных исследований:
К.х.н.: Маняшину А.О., Гиренко Е.Е., Охлобыстину А.О., Охлобыстиной А.В., Хохлову В.А.
Аспирантам: Колдаевой Ю.Ю., Васильевой Е.А.
За участие в проведении квантово-химических расчетов:
К.х.н. Пащенко К.П., аспиранту Арефьеву Я.Б.
За помощь в синтезе комплексов металлов:
Д.х.н. Сидорову А.А. (в.н.с. ИОНХ РАН им. Курнакова Н.С., г. Москва)
За участие в проведении ЭПР-исследований:
Д.х.н., проф. Белевскому В.Н. (МГУ им. М.В. Ломоносова, г. Москва)
К.х.н. Поддельскому А.И. (м.н.с. ИМХ РАН, г. Нижний Новгород)
СОКРАЩЕНИЯ
ЦВА – циклическая вольтамперограмма
ЭСП – электронный спектр поглощения
РФА – рентгенофлуоресцентный анализ
ЭПР – электронный парамагнитный резонанс
Меd – электромедиатор
Замечания и отзывы в двух экземплярах, заверенные печатью, просим направлять на имя Ученого секретаря диссертационного совета ДМ 307.001.04 по адресу:
414025, г. Астрахань, ул. Татищева, 16, гл.уч. корп.
Типография Астраханского государственного технического университета:
414025, Астрахань, Татищева, 16
Тираж 100. Заказ №……. от 2012
[1] Symons Martyn C.R. // Chem. Phys. – 1999. - Vol. - 20. N. 1. – P. 4767.
[2] А. с. 814273 СССР. Шим К. С., Скшек А. Е. – 1981. – Б. И. № 10.
[3] Ряшенцева М.А. // Успехи химии. – 1994. – Т.63. – В.5. – С. 456;
Дерягина Э. Н., Воронков М. Г. // ХГС. – 2000. – № 1. – С. 3.
[4] Губина Т.И., Лабунская В.И., Корниенко Г.К., Бородина Л.А., Харченко В.Г. // ХГС. – 1995. – №5. – С. 624.
[5] Шарипов И.А., Насыров Х.М., Шарипов А.Х., Мазитов М.Ф. // ЖОрХ. – 2000. – Т. 36. – Вып. 1. – С. 116.
[6] Сидоров А.А., Понина М.О., Нефедов С.Е., Еременко И.Л., Устынюк Ю.А., Лузиков Ю.Н. // Журн. нерган. химии. – 1997. – Т. 42. – С. 953.
[7] Ромахин А.С., Загуменов В.А., Никитин Е.В., Каргин Ю.М. // ЖОХ. – 1995. – Т. 65, Вып. 8. – C. 1321.
[8] Магдесиева Т.В., Бутин К.П. // Успехи химии. – 2002. – 71 (3). – C. 255.
[9] Смолянинов И.В., Охлобыстин А.О., Поддельский А.И., Берберова Н.Т., Еременко И.Л // Координационная химия. - 2011. - Т.37. - № 1. – С. 14.