

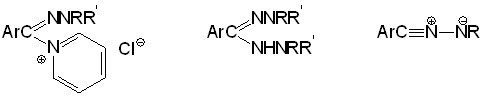
Синтез трихлорметиларенов и их реакции с пиридинами и другими нуклеофилами
На правах рукописи
ПОДДУБНЫЙ ИГОРЬ СЕРГЕЕВИЧ
СИНТЕЗ ТРИХЛОРМЕТИЛАРЕНОВ И ИХ РЕАКЦИИ С ПИРИДИНАМИ И ДРУГИМИ НУКЛЕОФИЛАМИ
02.00.03 – органическая химия
А В Т О Р Е Ф Е Р А Т
диссертации на соискание ученой степени
доктора химических наук
Саратов - 2009
Работа выполнена в лаборатории гетероциклических соединений Института органической химии имени Н.Д.Зелинского РАН, г.Москва и в научно-производственном центре ОАО «Каустик», г.Волгоград
Научный консультант:
Заслуженный деятель науки РФ,
доктор химических наук, профессор
Беленький Леонид Исаакович
Официальные оппоненты:
доктор химических наук, профессор Решетов Павел Владимирович
доктор химических наук, профессор Скворцов Игорь Михайлович
доктор химических наук, профессор Маркова Людмила Ивановна
Ведущая организация: Кубанский государственный технологический
университет
Защита состоится 15 октября 2009 г. в 14.00 часов на заседании диссертационного совета Д 212.243.07 при Саратовском государственном университете по адресу: 410012, Саратов, ул.Астраханская, 83, корпус 1, Институт Химии СГУ.
С диссертацией можно ознакомиться в Научной библиотеке Саратовского государственного университета им. Н.Г.Чернышевского
Автореферат разослан « 8 » сентября 2009 года
Ученый секретарь диссертационного совета Сорокин В.В.
ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ
Актуальность работы. Значение трихлорметиларенов (ТХМА) в синтетической органической химии определяется тем, что они являются удобными исходными веществами в синтезе карбоновых кислот и их производных, симметричных и несимметричных диарилкетонов и ряда гетероциклических систем, среди которых: 1,2,4- и 1,3,4-оксадиазолы, имидазолины, оксазолины, бензоксазолы, бензотиазолы, бензимида-золы, имидазолины, 1,3-оксазины, сим-триазины и 1,4-бензодиазепины. Относительная доступность и повышенная по сравнению с соответствующими карбоновыми кислотами реакционная способность бензотрихлоридов создают перспективу для их использования в качестве субстратов или интермедиатов не только в производстве крупнотоннажных органических соединений, например, таких как хлорангидриды бензойной, терефталевой, изофталевой и других кислот, но и в синтезе разнообразных биологически активных веществ. Восстановление трихлорметильной группы электрохимическим путем, либо под действием неорганических или органических реагентов, является одним из важных и перспективных путей синтетического использования ТХМА. Исследования в указанных направлениях, несомненно, имеют большое теоретическое и практическое значение и являются весьма актуальными, поскольку открывают возможности целенаправленного и эффективного применения трихлорметиларенов как в синтезе различных гетероциклов, так и для получения ароматических альдегидов и их разнообразных производных. Усовершенствование технологии получения ТХМА методом радикального хлорирования метилбензолов также имеет значительный практический потенциал, поскольку непосредственно связано с широким применением этих субстратов в синтезах различных промышленно ценных продуктов – ароматических кислот и их хлорангидридов, модификаторов резиновых смесей, пероксидных инициаторов и других соединений.
Цель работы. Установление закономерностей хлорирования метилбензолов и реакций трихлорметиларенов с пиридинами и другими нуклеофилами (гидразином, гидроксиламином, (тио)ацилгидразинами, водой, алифатическими и ароматическими кислотами, спиртами); установление механизма реакций ТХМА с нуклеофилами; выявление условий селективного получения ароматических и гетероциклических соединений, а также продуктов заданной степени хлорирования углеводородов.
Для достижения этой цели были сформулированы и решались следующие задачи:
- разработка общего способа селективного радикального хлорирования (галогени-рования) алкилароматических, алифатических и непредельных линейных углеводородов;
- выявление условий селективного и технически эффективного радикального хлорирования метилбензолов с получением продуктов заданной степени хлорирования;
- изучение механизма восстановительной конденсации трихлорметиларенов с гидроксиламином или гидразинами в присутствии пиридиновых оснований;
- выявление направлений и особенностей взаимодействия ТХМА с пиридинами, N-, O- и S-нуклеофилами в зависимости от строения реагентов и условий реакции;
- изучение реакций гетарилирования N- и С-нуклеофилов солями пиридиния, генерируемыми in situ из трихлорметиларенов и пиридинов;
- разработка препаративных методов синтеза симметричных и несимметричных 2,5-дизамещенных 1,3,4-окса(тиа)диазолов на основе реакций трихлорметиларенов с гидразином и (тио)ацилгидразинами;
- установление строения новых веществ методами спектроскопии ЯМР 1Н и 13С и масс-спектрометрии и выявление общих закономерностей и специфических особенностей в спектрах 2,5-дизамещенных 1,3,4-оксадиазолов.
Научная новизна. Впервые показаны общий характер и высокая эффективность стабилизирующего действия эфиров ортофосфорной кислоты в процессах радикального галогенирования метилароматических, предельных и непредельных линейных углеводо-родов. На примере хлорного железа и хлорида цинка определен диапазон концентраций кислот Льюиса, при которых стабилизирующее действие органических фосфатов обеспечивает селективное радикальное галогенирование углеводородных субстратов (метилбензолов) и получение высококачественных галогенированных продуктов.
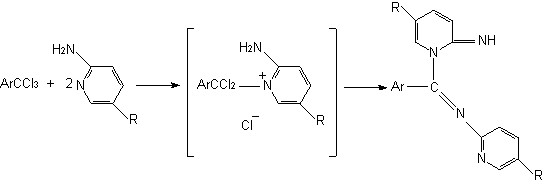
Установлено, что восстановительная конденсация трихлорметиларенов с гидразинами в пиридине протекает по двум конкурирующим направлениям, в одном из которых в качестве восстановителя выступает пиридин, а в другом – гидразин.
Первое направление включает образование N-(,-дихлорбензил)пиридиниевых солей, которые в зависимости от строения исходного трихлорметиларена претерпевают превращение либо в биспиридиниевые соли, либо в N-[N-(-хлорбензил)-4-пиридил]-пиридиниевые соли. Взаимодействие о,о’-дизамещенных трихлорметиларенов с пириди-ном в условиях восстановительной конденсации (при кипячении в пиридине) или в более мягких условиях (в хлороформе или хлористом метилене при обычной температуре) протекает с образованием N-[N-(-хлорбензил)-4-пиридил]пиридиниевых солей. Окисли-тельно-восстановительные превращения о,о’-дизамещенных ТХМА являются общими для пиридина, 3-R-замещенных пиридинов и хинолинов, причем в зависимости от нуклео-фильности атома азота реакция приводит к N-(-хлорбензил)-4-хлор-3-R-пиридиниевым солям, либо к N-[N(-хлорбензил)-3-R-4-пиридил]-3-R-пиридиниевым солям.
Предложена и обоснована общая схема реакций о,о’-дизамещенных ТХМА с пиридиновыми основаниями с восстановлением трихлорметильной группы и образова-нием ароматических альдегидов и их производных, что является новыми примерами окислительно-восстановительных превращений с участием аналогов никотинамидаденин-динуклеотида (НАДН) и его фосфата. Обнаружены и получены интермедиаты данной реакции: монопиридиниевые соли, 4-хлор-1,4-дигидроникотинамид, хлорид N-(-хлор-бензил)-4-хлорпиридиния и другие. Разработаны новые препаративные методы синтеза ароматических альдегидов, 4-хлорпиридинов и N-(4-пиридил)пиридиниевых солей.
Второе направление восстановительной конденсации реализуется для трихлор-метиларенов, не имеющих орто-заместителей или имеющих один орто-заместитель, и осуществляется при действии избытка гидразина на гидразоноилхлориды без участия пиридина в акте восстановления. Установлена принципиальная возможность реализации этого направления восстановительной конденсации и для трихлорметиларенов, имеющих два орто-заместителя, причем это направление является единственным, если восстановление через образование соответствующих пиридиниевых солей невозможно вследствие стерических препятствий.
Систематически исследованы конкурирующие направления взаимодействия трихлорметиларенов с N-, О- и S-нуклеофилами, не сопровождающиеся актом восста-новления. Впервые найдены оптимальные условия селективной гетероциклизации ТХМА под действием ацилгидразинов и гидразина. Разработаны простые и эффективные методы синтеза 2,5-дизамещенных 1,3,4-оксадиазолов и 1,3,4-тиадиазолов, исходя из трихлор-метиларенов и гидразидов карбоновых кислот или гидразингидрата. Впервые показано, что взаимодействие трихлорметиларенов и тиоацилгидразинов приводит к 2,5-дизамещен-ным 1,3,4-тиадиазолам с препаративными выходами.
Существенно расширена область применения ТХМА для синтеза ароматических альдегидов и их производных, гетероциклических систем (1,3,4-оксадиазолов и 1,3,4-тиадиазолов), в качестве «мягких» хлорирующих агентов для получения 4-хлорпиридинов и 1-(4-пиридил)пиридиниевых солей, а также в качестве алкилирующих реагентов для эффективной и -селективной активации пиридиновых оснований в реакциях гетарилиро-вания «жестких» и «мягких» нуклеофилов.
Выявлены общие закономерности и специфические особенности в спектрах ЯМР 1Н и 13С 2,5-дизамещенных 1,3,4-оксадиазолов. На основе спектров ЯМР 13С 1,3,4-оксади-азолов впервые проведена оценка электронного эффекта 5-R-1,3,4-оксадиазол-2-ильной группы как заместителя в бензольном кольце и количественная оценка трансмиссионной способности 1,3,4-оксадиазольного цикла.
Впервые выявлены и сформулированы общие закономерности в масс-спектрах электронного удара 2,5-дизамещенных 1,3,4-оксадиазолов и установлено, что метод масс-спектрометрии может использоваться для надёжной идентификации и доказательства их строения.
Практическая значимость работы. Разработаны технологичные и эффективные промышленные способы получения трихлорметиларенов, бензилидендихлоридов и бензилхлоридов путем селективного радикального хлорирования толуолов и ксилолов в присутствии фосфатных стабилизаторов. Эти способы позволяют получать целевые продукты заданной степени хлорирования с отличными выходами (до 99,5 %), высокой конверсией по хлору (до 99,8 %) и повышенной производительностью процесса.
В производственных условиях ОАО «Химпром», г.Волгоград впервые апроби-рованы или внедрены:
- способ получения,,,,,-гексахлор-пара-ксилола;
- способ получения бензилхлорида путем селективного радикального хлорирования толуола в присутствии фосфатных стабилизаторов;
- способ совместного получения ацетилхлорида и бензальдегида каталитическим взаимодействием бензилидендихлорида и уксусной кислоты;
- двухстадийный способ получения бензальдегида из кубовых остатков произ-водства хлористого бензила, содержащих бензилидендихлорид;
- способ получения и способ стабилизации жидких и твердых хлорированных парафинов (марок ХП-13, ХП-250, ХП-470, ХП-1100);
- способ получения нового многофункционального модификатора резиновых смесей на основе гексахлор-пара-ксилола – гексола М.
Установлено, что использование эфиров ортофосфорной кислоты в качестве эффективных дезактиваторов кислот Льюиса и стабилизаторов хлорированных (галогенированных) углеводородов на стадиях синтеза и выделения повышает выход и качество целевых продуктов, снижает их производственную себестоимость и расширяет их потребительский потенциал в процессах переработки и применения.
Разработаны удобные, одностадийные способы получения 2,5-дизамещенных 1,3,4-оксадиазолов, 1,4-фениленбис-1,3,4-оксадиазолов, 2,5-дизамещенных 1,3,4-тиадиазолов, на основе ТХМА и гидразина, гидразидов карбоновых, тиокарбоновых кислот.
Разработан метод получения о,о’-дизамещенных бензальдегидов и их азинов или гидразонов с высокими выходами с одновременным получением 3-R-замещенных 4-хлорпиридинов или N-(3-R-4-пиридил)-3-R-пиридиниевых солей. Впервые показано, что пиридиниевые соли, генерируемые in situ из о,о’-дизамещенных ТХМА и пиридина, выступают в качестве реакционноспособных агентов в реакциях селективного -гетарилирования различных N- или C-нуклеофилов. Это позволяет рассматривать о,о’-дизамещенные ТХМА в качестве удобных и специфических алкилирующих реагентов для эффективной и -селективной активации пиридиновых оснований в реакциях гетарилиро-вания «жестких» и «мягких» нуклеофилов.
Выполненные исследования существенно расширяют синтетический потенциал трихлорметиларенов в промышленном и препаративном синтезе различных соединений и химических продуктов: ароматических альдегидов и их производных, 4-замещенных пиридинов, 2,5-дизамещенных 1,3,4-оксадиазолов и 1,3,4-тиадиазолов, многофункцио-нальных модификаторов резиновых смесей и полимерной серы.
На защиту выносятся следующие положения:
- условия селективного и/или эффективного радикального хлорирования алкил-ароматических, предельных и непредельных линейных углеводородов с получением продуктов заданной степени хлорирования.
- закономерности и механизм реакции восстановительной конденсации трихлор-метиларенов с гидразином в пиридине;
- закономерности и механизм окислительно-восстановительных превращений о,о’-дизамещенных трихлорметиларенов под действием пиридиновых оснований;
- закономерности, механизм и особенности превращений трихлорметиларенов под действием двух или более нуклеофилов (N-, O- и S-нуклеофилов) с образованием гетероциклических систем, продуктов восстановления трихлорметильной группы или продуктов нуклеофильного замещения;
- реакционная способность пиридиниевых солей, генерируемых in situ из трихлор-метиларенов и пиридиновых оснований; закономерности превращения этих солей под действием нуклеофилов.
Апробация работы. Результаты исследований были представлены на Межвузовской конференции «Карбонильные соединения в синтезе гетероциклов» (Саратов, 1992), XVIII конференции по химии и технологии органических соединений серы (Казань, 1992), XIV Международном конгрессе по гетероциклической химии (Антверпен, 1993), XVI Международном симпозиуме по химии органических соединений серы (Мерзебург, 1994), X Международной конференции по органическому синтезу (Бангалор, 1994), Межинститутском коллоквиуме “Химия азотистых гетероциклов” (Черноголовка, 1995), XIV Международном конгрессе по гетероциклической химии (Тайбей, 1995), V Симпозиуме по гетероциклической химии «Голубой Дунай» (Часта Паперничка, 1995), VII Межвузовской конференции «Новые достижения в органической химии» (Саратов, 1997), Пятой Российской научно-практической конференции резинщиков “Сырье и материалы для резиновой промышленности. Настоящее и будущее” (Москва, 1998), 1-ой Всероссийской конференции по химии гетероциклов памяти А.Н.Коста (Суздаль, 2000), Электронной конференции по гетероциклической химии ECHET 98, Первой Международной конференции «Химия и биологическая активность азотистых гетероциклов и алкалоидов (Москва, 2001), XIV Международной научно-практической конференции резинщиков “Резиновая промышленность. Сырье. Материалы. Технологии” (Москва, 2008) и на XIX Симпозиуме «Проблемы шин и резинокордных композитов» (Москва, 2008).
Достоверность научных положений и выводов, приведенных в диссертационной работе, базируется на применении современных методов исследований и анализа (измерений), включая спектроскопию ЯМР 1Н и 13С, масс-спектрометрию, ИК- и УФ-спектроскопию, ВЭЖХ и газовую хроматографию, элементный анализ.
Публикации. По теме диссертации опубликовано 38 работ, из них 2 обзора и 13 статей в научных журналах, рекомендованных ВАК, 7 патентов на изобретения, 3 статьи в сборниках научных трудов и реферируемых журналах, 13 тезисов докладов на всерос-сийских и международных конференциях.
Личное участие автора заключалось в теоретическом обосновании тематики исследований, постановке задач, разработке теоретических положений, непосредственном участии во всех этапах исследовательских работ, а также в обработке, обобщении, интерпретации результатов исследований и формулировании выводов.
Структура и объём работы. Диссертационная работа изложена на 288 страницах машинописного текста, включая введение, 5 глав, выводы, список литературы из 299 наименований, 32 таблицы и 41 рисунок (схему). В первой главе приводятся и обсужда-ются результаты по синтезу и промышленному использованию трихлорметиларенов. Вторая глава посвящена обсуждению результатов исследования восстановительной конденсации трихлорметиларенов с гидразинами или гидроксиламином в пиридине. Третья глава посвящена изучению реакций N- и C-нуклеофилов с солями пиридиния, полученными из о,о’-дизамещенных трихлорметиларенов. Четвертая глава описывает результаты исследования реакций трихлорметиларенов с N-, O- и S-нуклеофилами, протекающих без восстановления трихлорметильной группы. Пятая глава представляет собой экспериментальную часть, в которой описаны методики синтеза соединений и выполнения измерений и анализа.
ОСНОВНОЕ СОДЕРЖАНИЕ РАБОТЫ
1. Синтез и промышленное использование трихлорметиларенов
Основным методом синтеза бензотрихлорида и его производных, содержащих инертные заместители, является исчерпывающее радикальное хлорирование одной или более метильных групп, инициируемое при помощи лабильных соединений (радикальных инициаторов) или светового излучения. Известно, что кислоты Льюиса, и в первую очередь соединения железа, присутствующие в исходных реагентах (смесях) в концентра-циях от 0,00001 мас.% до 0,001 мас.%, обусловливают обрыв цепей, снижают конверсию хлора, катализируют побочное электрофильное хлорирование метилбензолов в ядро и их алкилирование по Фриделю-Крафтсу промежуточно возникающими бензилхлоридами.
С целью снижения нежелательного каталитического эффекта кислот Льюиса в реакциях радикального хлорирования метилбензолов используют добавки органических или неорганических соединений, которые способны образовывать с кислотами Льюиса неактивные комплексы: пентаэритрит, полипропиленгликоль, маннит, триэтиламин, диизопропиламин, пиперидин, морфолин, диэтаноламин, мочевина, тиомочевина, N-замещенные амиды алифатических кислот, уротропин. Однако стабилизирующее действие ранее использовавшихся соединений-комплексообразователей в общем случае оказывается недостаточным для селективного радикального хлорирования метилбензолов и получения высококачественных и стабильных трихлорметиларенов.
В этой связи представлялось весьма актуальным выявить стабилизаторы, позволя-ющие осуществлять селективное радикальное хлорирование метилбензолов при относи-тельно высоком содержании кислот Льюиса в реакционной среде - более 0,00001 мас.%.
В рамках этой задачи нами был разработан способ селективного радикального хлорирования метилароматических углеводородов (1а-е) с инициированием процесса при помощи азобисизобутиронитрила (ABIBN) или УФ-облучения при температуре 80-130 °С в присутствии эфира ортофосфорной кислоты, взятого в количестве 0,1-1,0 мас.%, или его смеси с диметилформамидом (ДМФА) в массовом соотношении 1:1 (схема 1).
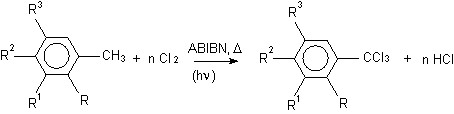
1а-е 2а-е 98,3-99,4 %, где n = 3 или 6
1, 2: R = R1 = R2 = R3 = H (а), R = Cl, R1 = R2 = R3 = H (б); R = R1 = R3 = H, R2 = Cl (в);
R = R2 = Cl, R1 = R3 = H (г); 1д: R = R2 = R3 = H, R1 = CH3; 1е: R = R1 = R3 = H; R2 = CH3;
2д R = R2 = R3 = H, R1 = CCl3; 2е R = R1 = R3 = H; R2 = CCl3;
СХЕМА 1
Использование эфира ортофосфорной кислоты (триалкилфосфата, диалкиларил-фосфата, алкилдиарилфосфата или триарилфосфата) в количестве 0,1-1,0 % от массы исходного метилбензола обеспечивает получение целевых ТХМА 2а-е с отличными выходами 98,3-99,4 %, с высокой конверсией хлора и селективностью, что подтвержда-ется высокой массовой долей ТХМА 2 в продуктах хлорирования - от 97,5 % до 99,4 %.
На примере процесса хлорирования п-ксилола впервые была проведена оценка стабилизирующего эффекта трибутилфосфата (ТБФ), производимого в промышленном масштабе, и его смеси с ДМФА (1:1) по сравнению со стабилизирующим действием других известных комплексообразователей.
Результаты этой части исследования приведены в таблице 1.
Таблица 1. Сравнительная эффективность стабилизаторов процесса
радикального хлорирования п-ксилола
| № п/п | Массовая доля железа в п-ксилоле, % | Стабилизатор | Массовая доля стаби-лизатора, % от п-ксилола | Выход ТХМА, % | Массовая доля основ-ного веще-ства, % | Избыток хлора, % от теории |
| 1 | 6•10-4 | уротропин | 0,2 | 98,2 | 98,5 | 43,4 |
| 2 | 6•10-4 | мочевина | 0,2 | 94,0 | 94,1 | 58,6 |
| 3 | 6•10-4 | тиомочевина | 0,2 | 95,7 | 95,5 | 53,7 |
| 4 | 6•10-4 | ДМФА | 0,2 | 98,8 | 98,8 | 26,2 |
| 5 | 6•10-4 | трибутилфосфат | 0,2 | 99,8 | 99,4 | 22,9 |
| 6 | 6•10-4 | ТБФ + ДМФА | 0,2 | 99,5 | 99,2 | 23,3 |
| 7 | 3•10-3 | уротропин | 0,4 | 96,9 | 96,3 | 82,8 |
| 8 | 3•10-3 | этаноламин | 0,4 | 94,9 | 95,1 | 94,2 |
| 9 | 3•10-3 | триэтаноламин | 0,4 | 95,4 | 95,0 | 93,6 |
| 10 | 3•10-3 | ацетамид | 0,4 | 95,3 | 95,5 | 92,0 |
| 11 | 3•10-3 | бензамид | 0,4 | 95,5 | 95,4 | 89,1 |
| 12 | 3•10-3 | ДМФА | 0,4 | 98,0 | 97,6 | 37,5 |
| 13 | 3•10-3 | пиридин | 0,4 | 97,1 | 96,5 | 87,4 |
| 14 | 3•10-3 | ТБФ | 0,4 | 99,2 | 99,0 | 30,8 |
| 15 | 3•10-3 | ТБФ + ДМФА | 0,4 | 99,2 | 98,9 | 31,7 |
| 16 | 5•10-3 | ТБФ + ДМФА | 0,4 | 99,0 | 98,7 | 35,9 |
Примечание: Хлорирование 0,25 моля п-ксилола осуществлялось в стандартных условиях - при 80-120 °С с использованием в качестве радикального инициатора ABIBN, взятого в количестве 1,1 % от массы п-ксилола, или при УФ-облучении.
Установлено, что традиционно используемые стабилизаторы радикального хлори-рования, как правило, проявляют положительный эффект только при относительно низких концентрациях кислот Льюиса, в частности при концентрациях ионов железа в исходном сырье (в хлоре и метилбензолах) – не более (1-5)•10-4 мас.%, в то время как ТБФ и его смесь с ДМФА оказывают заметный стабилизирующий эффект даже при такой высокой массовой доле железа в п-ксилоле, как 0,005 %.
Стабилизирующая роль эфиров ортофосфорной кислоты заключается в образова-нии стабильных, недиссоциирующих комплексов с солями железа и другими кислотами Льюиса, что нивелирует негативный ингибирующий эффект кислот Льюиса, присутст-вующих в реакционной среде, в радикальных процессах и подавляет побочное хлорирование ароматических субстратов в ядро. Образование устойчивых комплексов между алкил(арил)фосфатами и солями железа было подтверждено при помощи УФ-спектроскопии на примере смеси трибутилфосфата с хлорным железом (таблица 2).
Таблица 2. Данные электронных спектров поглощения ТБФ и комплекса FeCl3ТБФ
| Компонент или смесь | Длина волны,, нм | ||||||||||
| 300 | 310 | 320 | 330 | 340 | 350 | 360 | 370 | 380 | 390 | 400 | |
| Относительная оптическая плотность (по воздуху), % | |||||||||||
| ТБФ | 84,9 | 92,2 | 96,2 | 98,3 | 101,8 | 103,6 | 105,1 | 105,7 | 106,3 | 106,9 | 107,1 |
| ТБФ + 0,005 мас.% FeCl3 | 3,9 | 2,8 | 3,7 | 9,5 | 18,6 | 10,0 | 3,2 | 3,8 | 10,3 | 29,1 | 57,4 |
Как следует из данных таблицы 2, спектр поглощения ТБФ, содержащего 0,005 мас.% FeCl3, резко отличается от спектра поглощения «чистого» трибутилфосфата появлением новых полос в области 312 нм и 363 нм. Последняя полоса соответствует поглощению образующегося комплекса FeCl3ТБФ и практически совпадает с полосой поглощения комплекса FeCl3триэтилфосфат (364 нм), наблюдавшейся ранее в работе (Розенберг В.Р., Моцарев Г.В., Ушаков А.А., Суворов Б.А. Нефтехимия, 1974, т.14, № 6, С.885-890).
Высокая эффективность указанных фосфатных стабилизаторов позволяет осущест-влять процесс радикального хлорирования метилбензолов селективно даже при таком значительном содержании FeCl3 или хлорида цинка ZnCl2, как 0,01-0,03 мас.% (в пере-счете на массовую долю железа – 0,0034-0,01 %), что было наглядно продемонстрировано нами на примерах радикального хлорирования толуола (1а).
Таким образом, в исследованной нами серии метилароматических субстратов и стабилизаторов эффект дезактивации такой сильной кислоты Льюиса, как хлорное железо, оказался наиболее существенным именно для эфиров ортофосфорной кислоты, что, очевидно, связано с более высокой стабильностью и каталитической инертностью их комплексов с кислотами Льюиса.
1.2. Улучшенный метод радикального галогенирования алкилароматических, непредельных и предельных углеводородов
Используемые для получения ТХМА 2а-е стабилизаторы – эфиры ортофосфорной кислоты или стабилизирующая система органический фосфат – ДМФА – были приме-нены в процессах получения и выделения (ректификации или дистилляции) продуктов неполного радикального хлорирования толуола: бензилхлорида и бензилидендихлорида, а также их 2-хлор-, 4-хлор- и 2,4-дихлорзамещенных (схема 2):
ArCH3 + Cl2 ArCH2Cl + ArCHCl2 + ArСCl3 + HCl
1а-г 3а-г 4а-г 2а-г
1, 2, 3, 4: Ar = C6H5 (а), Ar = 2-ClC6H4 (б), Ar = 4-ClC6H4 (в), Ar = 2,4-Cl2C6H3 (г).
СХЕМА 2
Применение указанных стабилизаторов в количестве 0,05-0,5 % от массы исходного толуола позволяет увеличить выход целевых продуктов на 2-8 % и повысить конверсию хлора на 1-6 % по сравнению с процессами, в которых использовались другие известные стабилизаторы, например, уротропин, ацетамид, N,N-диметилформамид, мочевина, триэтаноламин. Эти результаты также свидетельствуют о том, что эфиры ортофосфорной кислоты более эффективно ингибируют каталитические свойства кислот Льюиса нежели другие апробированные стабилизаторы-комплексообразователи.
Выполненные исследования позволили разработать модифицированный способ получения бензилхлоридов и бензилидендихлоридов, характеризующийся высокой селективностью процесса, высокими выходами и качеством целевых продуктов и более высокой конверсией по хлору по сравнению с другими промышленными и препаративными способами получения. Этот способ был успешно апробирован нами в производственных условиях ОАО «Химпром», г.Волгоград на примере получения и выделения высококачественного хлористого бензила.
Учитывая полученные результаты радикального хлорирования метилбензолов 1 с получением ТХМА 2, бензилхлоридов 3 и бензилидендихлоридов 4, мы предположили, что эффективность стабилизирующего действия эфиров ортофосфорной кислоты носит общий характер и распространяется также на реакции радикального хлорирования других углеводородных субстратов – алканов и алкенов. Как известно, эти реакции также весьма чувствительны к содержанию кислот Льюиса, которые не только ингибируют радикальный процесс, уменьшают конверсию хлора, но и существенно снижают термическую стабильность и качество целевых продуктов – хлорированных парафинов по показателям «массовая доля кислот в пересчета на HCl» и «цветность по иодной шкале».
Высказанное предположение было наглядно подтверждено нами сначала на примере успешного получения твердых хлорированных парафинов с массовой долей хлора в пределах 70-72 %, а затем и на примерах получения высокостабильных галогенированных парафинов с массовой долей галогена в пределах 12-72 %.
В качестве исходных углеводородных субстратов использовали жидкие и твердые парафины различных фракций с длиной углеродной цепи в пределах C14-C38, а также различные промышленные фракции -олефинов с длиной углеродной цепи в пределах C12-C38. В качестве галогенирующих агентов использовали газообразный хлор и жидкий бром. Инициирование радикального процесса осуществляли при помощи УФ-облучения либо с использованием азобисизобутиронитрила.
Радикальное галогенирование исходных углеводородов проводили по одно-, двух- или трехреакторной каскадной схеме с противотоком реагентов при температуре в пределах 70-155 °С (в зависимости от природы галогена и необходимой степени галогенирования) в присутствии 0,05-2,0 мас.% эфира фосфорной кислоты (триалкил-фосфата, диалкиларилфосфата, алкилдиарилфосфата, триарилфосфата или их смеси).
Результаты получения галогенированных парафинов свидетельствуют, что эфиры фосфорной кислоты, применяемые в качестве стабилизаторов процесса галогенирования алканов и алкенов, проявляют заметно более выраженный стабилизирующий эффект по сравнению с другими органическими соединениями - ДМФА, уротропином, триэтанол-амином и тем более по сравнению с радикальным процессом, осуществляемым без какого-либо стабилизатора. Так, в производственных условиях ОАО «Химпром» при получении жидких хлорпарафинов марок ХП-250 и ХП-470 было установлено, что введение на стадию хлорирования 0,10-0,15 мас.% трибутилфосфата позволяет уменьшить проскок хлора на 10-20 % и сократить общую продолжительность стадии отдувки кислых примесей на 25-30 %, что соответственно снижает энергетические затраты на процесс, повышает его производительность и улучшает качество целевых хлорпарафинов по показателям «цветность» и «массовая доля кислот в пересчете на HCl».
Выполненные исследования завершились разработкой нового эффективного способа радикального галогенирования предельных и непредельных углеводородов, отличающегося высокой конверсией галогена и повышенной производительностью процесса. Разработанный способ позволяет получать высококачественные, термически стабильные галогенированные парафины, применяемые в нефтехимической отрасли, в производстве шин, РТИ и других разнообразных полимерных композиций и компаундов.
Обобщая результаты этого этапа исследований, можно констатировать, что нами разработан универсальный и эффективный метод радикального галогенирования алкилароматических, предельных и непредельных углеводородов в присутствии активно действующих органических стабилизаторов, обладающих высокой комплексообразующей способностью по отношению к кислотам Льюиса, - эфиров ортофосфорной кислоты. Определена область практического применения алкил(арил)фосфатов в зависимости от природы исходного углеводородного субстрата, содержания в нем кислот Льюиса (в первую очередь, соединений железа) и заданной степени галогенирования:
- селективное и эффективное радикальное галогенирование метилароматических субстратов достигается при массовой доле ионов железа (в общем случае – кислот Льюиса) – не более 10-3 % и при использовании 0,05-0,5 мас.% триалкилфосфата, диалкиларилфосфата, алкилдиарилфосфата, триарилфосфата или их смесей с ДМФА;
- эффективное радикальное галогенирование алканов и алкенов с получением высококачественных и стабильных галогенированных парафинов обеспечивается при использовании субстратов с массовой долей железа (кислот Льюиса) не более 0,01 % и при использовании триалкилфосфата, диалкиларилфосфата, алкилдиарилфосфата, или триарилфосфата, взятого в количестве 0,05-5,0 % от массы исходного субстрата.
Таким образом, нами показано, что применение эфиров фосфорной кислоты в качестве дезактиваторов кислот Льюиса и стабилизаторов в процессах радикального галогенирования алкилароматических, предельных и непредельных углеводородов и в процессах выделения и стабилизации их галогенированных производных позволяет:
- повысить выход целевых хлорированных продуктов с 97,0 % до 98,4-100 %;
- повысить массовую долю основного вещества в целевых продуктах (ТХМА) с 95,0-97,5 % до 98,4-99,4 %;
- увеличить конверсию хлора с 94,5 % до 99,8 % (при каскадной схеме хлорирования) или снизить избыток хлора на 10-20,5 % (при однореакторной схеме хлорирования);
- увеличить производительность технологического оборудования (на единицу реакционного объема) на 9-18 %;
- существенно улучшить ряд технических характеристик (показателей) целевых хлорорганических продуктов – «цветность» (снижение цветности на 1-9 единиц по иодной шкале), «термостабильность» (улучшение термостабильности на 26-40 %), «массовая доля кислот в пересчете на HCl» (снижение кислотности на 50-90 % или в 2-10 раз), что заметно повышает качество и потребительский потенциал получаемых продуктов;
- использовать углеводородные субстраты с более высокой массовой долей железа (вплоть до 0,01 % для алканов и алкенов и до 10-3 % для метилароматических субстратов), что значительно расширяет сырьевую базу производства хлорированных (галогениро-ванных) углеводородов;
- упростить технологию производства за счет исключения стадии предварительной очистки углеводородных субстратов от примесей железа (кислот Льюиса) и/или стадии очистки целевых продуктов от нежелательных примесей и соответственно снизить себестоимость целевых галогенорганических продуктов;
- сократить общее количество сточных вод и отходов процесса и снизить техноген-ное воздействие производства на окружающую среду.
1.3. Практическое использование трихлорметиларенов и продуктов неполного радикального хлорирования метилбензолов
Трихлорметиларены 2д и 2е с двумя трихлорметильными группами широко используются в промышленном органическом синтезе для получения различных хлорорганических продуктов. Так, известно применение ГХПК (2е) в качестве активно действующего вещества для получения различных композиционных, многофункциональ-ных модификаторов резиновых смесей на основе ненасыщенных каучуков. Модифици-рующие свойства ГХПК основаны на его способности на первом этапе производства замедлять подвулканизацию резиновых смесей (т.е. выступать в качестве антискорчинга), а на втором этапе - ускорять их вулканизацию совместно с традиционными ускорителями вулканизации и заметно улучшать физико-механические характеристики целевых резин.
Синтезированный нами гексахлор-п-ксилол (2е) без дополнительной очистки был использован в качестве активно действующего компонента при разработке новых оригинальных многофункциональных модификаторов для резиновых смесей. Так, в рамках исследования были разработаны и запатентованы два варианта многокомпонент-ного модификатора «Гексол М», а также разработана оригинальная технология его получения, отличающаяся простотой и экономичностью и характеризующаяся низким сырьевым индексом и минимальным негативным воздействием на окружающую среду.
Эффективность действия разработанных модификаторов была оценена в условиях ОАО «Волтайр», г.Волжский (Волжский шинный завод) при получении резиновых смесей на основе 100 массовых частей каучука СКИ-3, применяющихся в настоящее время на шинных заводах при производстве радиальных шин.
Резины, содержащие новый модификатор, превосходят резину с серийным модификатором - гексолом ХПИ (ТУ 2471-005-00209906-94), взятым в той же дозировке, по следующим показателям: клейкости - на 22-66 кПа; уровню условного напряжения при удлинении 300 % - на 0,4-1,2 МПа; сопротивлению раздиру - на 4-7 кН/м; прочности связи резины с кордом 23 КНТС - на 18-29 Н. По остальным показателям резины с опытными образцами модификатора либо равноценны, либо превосходят резину с серийным модификатором гексолом ХПИ.
Разработанная нами технология получения нового модификатора «гексол М» была успешно апробирована в производственных условиях ОАО «Химпром», г.Волгоград путем выпуска опытно-промышленной партии модификатора (ТУ 2471-252-05763458-98), которая была успешно испытана в условиях промышленного производства в ОАО «Волтайр», г.Волжский при выпуске радиальных шин. Было установлено, что гексол М по своим потребительским и техническим свойствам не уступает другим выпускаемым в промышленном масштабе многофункциональным модификаторам на основе гексахлор-п-ксилола – гексолу ЗВИ (ТУ 2471-007-00209906-95), гексолу ХПИ (ТУ 2471-005-00209906-94) и гепсолу-ХКП (ТУ 6-01-5-81-97, СТО 00203275-220-2008), а по некоторым показателям целевых резин превосходит указанные модификаторы.
Еще одним направлением практического использования трихлорметиларенов стало их успешное применение в качестве соединений, способных инициировать и поддержи-вать термическую полимеризацию циклооктасеры S8, регулировать рост полимерной цепи серы, стабилизировать её концевые фрагменты и препятствовать реверсии полимерной серы в циклооктасеру.
На примере гексахлорпараксилола был продемонстрирован эффект стабилизации полимерной серы, заключающийся во взаимодействии концевых атомов серы полимерной цепи с трихлорметильной группой, с атомом хлора или с дихлорбензильным радикалом с образованием производных серы, например, сульфенилхлоридов:
S-(S)n-S + Cl3C-C6H4-CCl3 Cl3C-C6H4-CCl2-S-(S)n-SCl
2(S-(S)n-S) + Cl3C-C6H4-CCl3 ClS-(S)n-S-Cl2C-C6H4-CCl2-S-(S)n-SCl
S-(S)n-S + 2Cl3C-C6H4-CCl2 Cl3C-C6H4-CCl2-S-(S)n-S-Cl2C-C6H4-CCl3
Использование ГХПК в качестве стабилизатора полимерной серы позволяет не только повысить выход целевого продукта с 35-37 % (без стабилизатора) до 42-46 %, но и обеспечить его высокую стабильность при хранении за счет предотвращения реверсии полимерной серы в циклическую форму.
Выполненные нами исследования легли в основу разработанного и запатенто-ванного способа получения полимерной серы, в котором ключевыми стадиями процесса являются стадии термической полимеризации серы, стабилизации цепи гексахлор-пара-ксилолом 2е и закалки (резкого охлаждения) полимеризата.
В качестве инициаторов полимеризации циклооктасеры и стабилизаторов полимер-ной серы могут использоваться и другие ТХМА 2, в первую очередь, бифункциональный гексахлор-мета-ксилол (2д) и высоко реакционноспособные о,о'-диметилзамещенные бензотрихлориды - 2,4,6-триметилбензотрихлорид (2и), 2,3,5,6-тетраметил- и 2,3,4,6-тетраметилбензотрихлориды (2к) и (2л).
В результате исследований расширена область практического применения ТХМА 2 в химической, шинной и резинотехнической отрасли промышленности путем создания и применения оригинальных рецептур и экологически эффективных технологий получения высококачественного многофункционального модификатора резиновых смесей «гексола М» и получения «невыцветающего» вулканизующего агента - полимерной серы.
2. Восстановительная конденсация трихлорметиларенов с гидразинами в пиридине.
2.1. Влияние соотношения реагентов на состав и природу продуктов
восстановительной конденсации
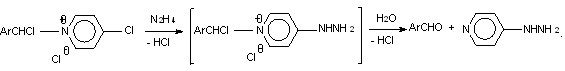
Ранее в лаборатории гетероциклических соединений ИОХ РАН было обнаружено, что взаимодействие трихлорметиларенов (ТХМА) 2 с гидроксиламином или гидразинами в пиридине приводит к ряду продуктов восстановительной конденсации: к оксимам 10 и нитрилам 11 (образуются при дегидратации оксимов 10) или к бензальдазинам 13 и гидразонам 16.
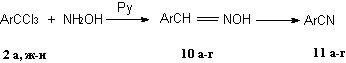

10, 11, 13: а Ar = C6H5; б Ar = 2,4-Me2C6H3; в Ar = 2,4,5-Me3C6H2; г Ar = 2,4,6-Me3C6H2
СХЕМА 3
В результате восстановительной конденсации бензотрихлорида (2а) с гидразином, наряду с продуктом восстановления трихлорметильной группы - бензальдазином (13а), был также выделен с небольшим выходом 2,5-дифенил-1,3,4-оксадиазол (14а).
Казалось очевидным, что роль восстановителя в этих превращениях играет гидроксиламин или гидразин, использовавшиеся в большом (5-10-кратном) избытке, однако попытки восстановить указанными реагентами соответствующие гидроксимоил- и гидразоноилхлориды, которые рассматривались в качестве наиболее вероятных интермедиатов, не дали чётких результатов – восстановление наблюдалось не во всех случаях. Оставалось неясным, на какой стадии идет восстановление трихлорметильной группы, и какова роль пиридина в этих реакциях.
Для выяснения этих вопросов нами было исследовано влияние молярного соотношения реагентов на природу и состав продуктов восстановительной конденсации. Так, было показано, что уменьшение избытка гидроксиламина вплоть до эквимолярного соотношения с ТХМА 2а,ж-и не приводит к существенному снижению выходов продуктов восстановления – оксимов 3. Аналогичные результаты были получены нами при варьировании количества гидразина в реакциях с ТХМА 2а,ж-и: при уменьшении количества гидразина основными продуктами реакции остаются соответствующие бензальдазины 13а-г, а в случае мезитотрихлорида 2и выход азина 13г при эквимолярном количестве гидразина составил ~ 50 %. Неожиданным оказалось образование небольших количеств продуктов, которые по данным ИК-, ПМР-спектроскопии, масс-спектрометрии и элементного анализа были идентифицированы как не описанные ранее 4-пиридил-гидразоны замещенных бензальдегидов 20б-г.
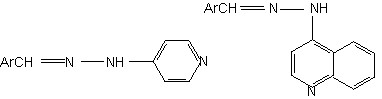
20б-г 5-16 % 21 20 %
20: Ar = 2,4-Me2C6H3 (б); Ar = 2,4,5-Me3C6H2 (в); 20г, 21 Ar = 2,4,6-Me3C6H2
Аналогичный продукт – 4-хинолилгидразон мезитоальдегида 21 (выход 20 %) был выделен и при взаимодействии мезитотрихлорида 2и с гидразином в хинолине.
Таким образом, пиридин (хинолин) в процессе восстановительной конденсации является не только растворителем и основанием, связывающим выделяющийся хлороводород, а сам участвует в окислительно-восстановительных превращениях. Такая роль пиридина естественно объясняла и отмеченные выше хорошие выходы оксима 10г и альдазина 13г при эквимольных соотношениях трихлорида 2и с гидроксиламином и гидразином. Дальнейшее исследование позволило выявить основные ступени механизма обсуждаемой реакции.
2.2 Природа восстановителя и механизм восстановительной конденсации
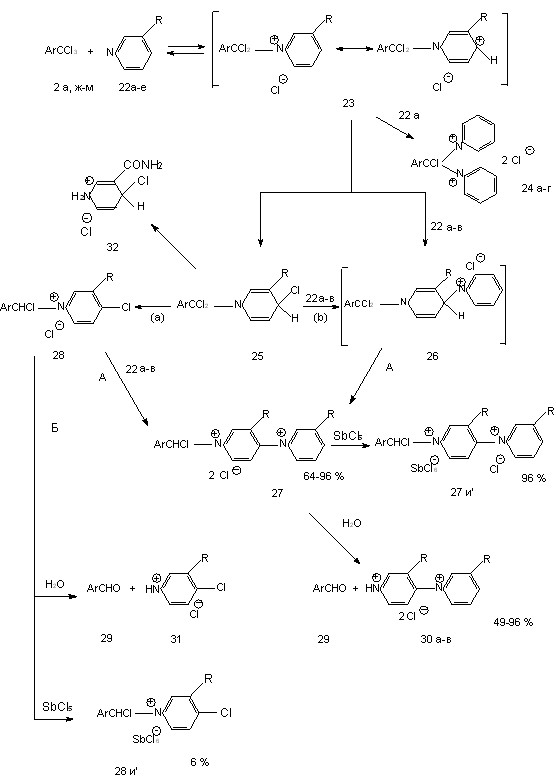
о,о’-дизамещенных трихлорметиларенов с гидроксиламином или гидразинами в пиридине
Полагая, что пиридин непосредственно участвует в восстановлении трихлор-метильной группы, мы исключили из реакционной среды гидроксиламин и гидразин и исследовали непосредственное взаимодействие ТХМА 2а,ж-м с избытком пиридина в растворе хлороформа или хлористого метилена. Было показано, что первой ступенью взаимодействия между пиридином 22а и ТХМА 2а,ж-м является образование хлоридов 1-(,-дихлорбензил)пиридиния 23 (схема 4). Эти соли под действием второй молекулы пиридина могут претерпевать нуклеофильное замещение подвижного -атома хлора дихлорметиленовой группы с образованием дихлорида 1,1’-(-хлорбензил)биспиридиния (24). Согласно литературным данным, подобное направление реакции легко реализуется для родственных N-(-галогеналкил)пиридиниевых солей, приводя к соответствующим биспиридиниевым солям. Нами впервые показано, что такое превращение характерно для монопиридиниевых солей 23, полученных из ТХМА 2а,ж,з,м, но не из стерически затрудненных о,о-дизамещенных ТХМА 2и-л, (схема 4).
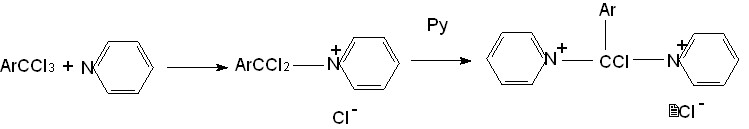 2а,ж-м 22а 23а,ж-м 24а,ж,з,м 58-93 %
2а,ж-м 22а 23а,ж-м 24а,ж,з,м 58-93 %
2, 23, 24: Ar = Ph (a), Ar = 2,4-Me2C6H3 (ж); Ar = 2,4,5-Me3C6H2 (з); Ar = 2,4,6-Me3C6H2 (и);
Ar = 2,3,4,6-Me4C6H (к); Ar = 2,3,5,6-Me4C6H (л); Ar = 2,3,4,5-Me4C6H (м).
СХЕМА 4
Как известно, образование солей 23а и 24а рассматривалось в качестве начальных стадий реакции Фудживары, протекающей при взаимодействии бензотрихлорида 2а с пиридином и водной щелочью, однако не было экспериментально подтверждено.
Впервые полученные с хорошими выходами (58-93 %) биспиридиниевые соли 24а,ж,з были охарактеризованы спектрами ЯМР 1Н. 2-Метил-, 2,3-диметил- и 2,6-диметилпиридины и 8-метилхинолин в этих же условиях не вступают в реакцию с трихлоридами 2ж и 2з, очевидно, вследствие стерических препятствий. Взаимодействие бензотрихлорида с пиридином в хлороформе не идет даже при нагревании, но происходит при кипячении бензотрихлорида 2а в избытке пиридина, причем основным продуктом реакции является биспиридиниевая соль 24а. Монопиридиниевую соль 23а удается получить в виде стабильного гексахлорантимоната 23а (~ 50 %) при добавлении эквивалентного количества пиридина к предварительно полученной суспензии соли PhCCl2+SbCl6 в хлористом метилене.
о,о-Диметилзамещенные ТХМА 2и-л не образуют биспиридиниевых солей типа 24, очевидно, из-за стерических препятствий. Эти ТХМА, несомненно, способны к образованию монопиридиниевых солей типа 23, о чем свидетельствует успешное получение нами такой соли (23и) из мезитотрихлорида 2и и 4-пиколина (86 %). Однако, подобные соли со свободным положением 4 пиридинового цикла выделить не удается, так как они вступают во взаимодействие с пиридином, которое может приводить не к биспиридиниевым солям 24, а к принципиально иным продуктам, указанным ниже.
Другой тип превращений монопиридиниевых солей 23, представленный на схеме 5, заключается в нуклеофильной атаке хлорид-анионом или пиридином стерически незатрудненного электронодефицитного положения 4 пиридиниевого цикла, что приводит к образованию N-замещенного 4-хлор-1,4-дигидропиридина 25 или N-замещенного 4-(1-пиридинио)-1,4-дигидропиридина 26. Ароматизация последнего с восстановлением одного из бензильных атомов хлора может привести к N-(4-пиридил)пиридиниевой соли 27. Еще один возможный путь образования соли 27 – ароматизация N-замещенного 1,4-дигидропиридина 25 до 4-хлорпиридиниевой соли 28 и превращение последней в соль 27. Отметим, что синтез 1-(4-пиридил)пиридиниевых солей из 4-галогенпиридинов известен.
Установлено, что при проведении реакции о,о’-дизамещенных бензотрихлоридов 2и-л с двумя молями пиридина в хлороформе или хлористом метилене в отсутствие гидроксиламина или гидразинов с хорошими выходами образуются соли 27и-л.
Строение N-(4-пиридил)пиридиниевых солей 27и-л подтверждено спектрами ЯМР 1Н в сухом ДМСО-d6, которые хорошо согласуются с литературными данными для N-(-хлоралкил)- и N-(-хлорарилметил)пиридиниевых солей (Anders E., Markus F., Meske H., Tropsch J.G., Maas G. // Chem. Ber., 1987, Bd. 120, S.735-745), а для соли 27и – также данными спектроскопии ЯМР 13С.
Гидролиз солей 27и-л в водном этаноле или водном ДМСО приводит с высокими выходами (до 95 %) к соответствующим замещенным бензальдегидам 29г-е и дихлориду N-(4-пиридил)пиридиния 30а. Гидролизаты гладко реагируют с гидроксиламином или гидразинами, образуя соответствующие производные бензальдегидов 10, 13 или 16 с выходами 60-90 %.
Аналогичные результаты дает и взаимодействие трихлорида 2и с пиридином в мольном соотношении 1 : 4, приводящее с высоким выходом (> 90 %) к соли 27и. Это свидетельствует о том, что подвижный атом хлора бензильного фрагмента соли 27и не может быть замещен находящимся в избытке пиридином из-за очевидных стерических препятствий. Взаимодействие мезитотрихлорида 2и с пиридином в эквимолярном соотношении приводит к получению соли 27и с выходом 76 % в расчете на пиридин, причем в продуктах гидролиза реакционной смеси обнаружены 2,4,6-триметил-бензальдегид 29и, дихлорид 1-(4-пиридил)пиридиния 30а, непрореагировавший исходный трихлорид 2и, а также незначительные количества гидрохлорида пиридина и 2,4,6-триметилбензойной кислоты.
2: a Ar = Ph, ж Ar = 2,4-Me2C6H3, з Ar = 2,4,5-Me3C6H2, и Ar = 2,4,6-Me3C6H2,
к Ar = 2,3,4,6-Me4C6H, л Ar = 2,3,5,6-Me4C6H, м Ar = 2,3,4,5-Me4C6H.
22: a R = H, б R = Me, в R = OH, г R = CONH2, д R = COOEt, e R = Br.
24: a Ar = Ph, б Ar = 2,4,-Me2C6H3, в Ar = 2,4,5-Me3C6H2, г 2,3,4,5-Me4C6H.
28и’: R = H, Ar = = 2,4,6-Me3C6H2; 30: а R = H, б R = Me, в R = OH.
31: а R = H, г R = CONH2, д R = COOEt, e R = Br.
СХЕМА 5
Образование осадка соли 27и, малорастворимой в CDCl3 или CD2Cl2, наблюдается уже через 2-3 ч после смешения реагентов, а концентрации интермедиатов 23, 25 и 26, вероятно, настолько малы, что удается обнаружить помимо исходных соединений лишь до 8 мас.% соли 27и. Можно предположить, что лимитирующей стадией реакции ТХМА с пиридином является образование монопиридиниевых солей 23, а последующие стадии протекают значительно быстрее. Добавление пентахлорида сурьмы к реакционной смеси в процессе взаимодействия трихлорида 2и с пиридином позволило зафиксировать в виде гексахлорантимоната 28и 4-хлорпиридиниевую соль, что является убедительным доводом в пользу протекания реакции через образование соединений 25 и 28. При этом возможность образования соли 28и через 1-(4-пиридил)пиридиниевую соль типа 27 исключается, поскольку соль 27и при взаимодействии со SbCl5 гладко превращается в соответствующий гексахлорантимонат 27и. Кроме того, если при взаимодействии ТХМА 2и с пиридином в хлороформе подвергнуть реакционную смесь гидролизу на ранних стадиях реакции, то по спектру ЯМР 1Н, наряду с пиридилпиридиниевой солью 30а, удается обнаружить 4-хлорпиридин 31а, а также гидрохлорид пиридина 22а•HCl, причем массовое соотношение продуктов 30а : 31а : 22а•HCl составляет 4:1:1.
Соли типа 27 были получены с хорошими выходами лишь для о,о-дизамещенных ТХМА 2и-л, однако такие соли удалось обнаружить и для стерически незатрудненных трихлоридов 2а,ж,з. Так, из продуктов взаимодействия трихлорида 2ж с пиридином в условиях, аналогичных использованным при синтезе солей 27и-л, наряду с биспиридиниевой солью 24ж, был выделен (после гидролиза) с выходом 4 % дихлорид N-(4-пиридил)пиридиния 30а. Бензотрихлорид 2а, при обычной температуре довольно инертный по отношению к пиридину, вступает в реакцию при кипячении в избытке последнего, т.е. в условиях восстановительной конденсации, причем наряду с биспиридиниевой солью 24а было выделено небольшое количество (6 %) дихлорида 1-(4-пиридил)пиридиния 30а. Эти результаты подтверждают, что в отсутствие стерических затруднений нуклеофильное замещение атома хлора в соли 23 осуществляется значительно быстрее, чем атака хлор-анионом или пиридином положения 4 этой соли, а также объясняют снижение выходов продуктов восстановительной конденсации (азинов, оксимов) при переходе от мезитотрихлорида к моно-орто-метилзамещенным ТХМА 2ж,з,м и бензотрихлориду 2а.
В отсутствие дополнительных стерических препятствий образование N-(4-пиридил)пиридиниевых солей 27 можно ожидать при взаимодействии тех же трихлоридов 2и-л с замещенными пиридинами, нуклеофильность или сила которых как оснований не ниже, чем у пиридина 22а, например, с 3-замещенными пиридинами 22б,в, несущими электронодонорные заместители. И действительно, нами было показано, что 3-метил-пиридин 22б и 3-гидроксипиридин 22в образуют с ТХМА 2и соли 27, которые при гидролизе дают 2,4,6-триметилбензальдегид 29и и дихлориды N-(3-R-пиридил-4)-3-R-пиридиния 30б,в (схема 5, путь А).
С целью обнаружения или выделения 1,4-дигидропиридиновых интермедиатов типа 25 или 26 мы использовали в исследуемой реакции 3-R-замещенные пиридины с электроноакцепторными заместителями, повышающими электрофильность положения 4 пиридинового цикла и стабилизирующими 1,4-дигидропиридиновую систему. При исследовании методом ЯМР 1Н продуктов реакции трихлорида 2и с никотинамидом 22г был зафиксирован гидрохлорид 4-хлор-1,4-дигидроникотинамида (32). Ароматизация соответствующего указанному 1,4-дигидропиридину интермедиата 25 с восстановлением,-дихлорметиленовой группы приводит после гидролиза к гидрохлориду 4-хлор-никотинамида 31г и 2,4,6-триметилбензальдегиду 29и (схема 5, путь «Б» и схема 6).
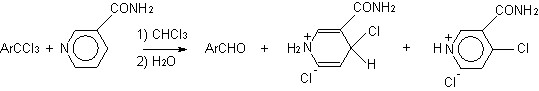
2и 22г 29и 32 31г•HCl
СХЕМА 6
Аналогично протекает и взаимодействие мезитотрихлорида 2и с этилникотинатом 22д, которое гладко приводит после гидролиза с выходами 60-65 % к 2,4,6-триметил-бензойному альдегиду (29и) и этиловому эфиру 4-хлорникотиновой кислоты (31д). Следует отметить, что в обоих рассмотренных случаях не были обнаружены соответствующие N-(4-пиридил)пиридиниевые соли типа 30, даже если в реакциях использовалось двойное молярное количество никотинамида или этилникотината. Такой результат легко объяснить с учетом как стерических препятствий атаке положения 4, так и пониженной (по сравнению с незамещенным пиридином) нуклеофильности никотинамида и 3-этоксикарбонилпиридина. Приведенные соображения согласуются и с результатами взаимодействия трихлорида 2и с 3-бромпиридином (22е), которое после гидролиза приводит к 3-бром-4-хлорпиридину (31е) и альдегиду 29и (выходы продуктов около 65%):
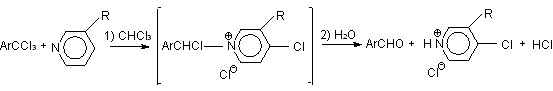
2и 22д,е 28 29и 31д,е 60-65 %
22, 28, 31: R = COOEt (д); R = Br (е)
СХЕМА 7
Из полученных результатов следует, что перенос водорода с 1,4-дигидропириди-нового цикла на дихлорметиленовую группу происходит не в 1,4-дигидропиридил-пиридиниевых солях типа 26, а в N-замещенных 4-хлор-1,4-дигидропиридинах 25. Последние (после ароматизации) могут превращаться в соли типа 27 при отсутствии стерических затруднений и достаточной нуклеофильности пиридинового основания, как это имеет место для 3-пиколина (22б) и 3-гидроксипиридина (22в). В случае пиридинов 22г-е с более объемными заместителями в положении 3 (CONH2, COOEt, Br), которые к тому же понижают нуклеофильность атакующего пиридина, взаимодействие (схема 5, путь а и схема 7) останавливается на стадии образования солей N-(-хлорбензил)-4-хлорпиридиния 28. Последние при гидролизе превращаются в замещенный бензальдегид 29 и 3-R-замещенные 4-хлорпиридины 31г-е, а при действии гидроксиламина или гидразинов – в соответствующие продукты восстановительной конденсации – оксимы и азины или гидразоны.
Важнейшей стадией процесса, определяющей все последующие превращения, является присоединение хлорид-аниона по положению 4 пиридиниевого катиона 23. Подобная атака хлорид-иона предполагалась как одна из ступеней механизма известного синтеза дихлорида N-(4-пиридил)пиридиния 30а из пиридина и хлористого тионила. Постулировалось, что первоначально образующийся хлорид N-(хлорсульфинил)-пиридиния превращается в 4-хлор-1-хлорсульфинил-1,4-дигидропиридин, который атакуется пиридином с замещением атома хлора и образованием 4-(1-пиридинио)-1,4-дигидропиридина типа 26, а последний в результате ароматизации дает 1-(4-пиридил)-пиридиниевую соль 30а. Следует отметить, что в литературе был рассмотрен и альтернативный механизм образования дихлорида 4-пиридилпиридиния 30а через ароматизацию 4-хлор-1-хлорсульфинил-1,4-дигидропиридина и нуклеофильное замеще-ние пиридином атома хлора в возникающей 4-хлорпиридиниевой соли. В нашей работе такой механизм впервые нашел прямое экспериментальное подтверждение, о чем свидетельствует обнаружение и выделение 1,4-дигидропиридина 32, 4-хлорпиридинов 31 и 4-хлорпиридиниевой соли 28и.
Ключевой стадией восстановительной конденсации о,о-дизамещенных ТХМА 2и-л, на которой собственно и происходит восстановление, является формальный перенос гидрид-иона (26 27 в маршруте А или 25 28 в маршруте а). Сам факт переноса водорода с пиридинового кольца на бензильный атом углерода подтвержден тем, что при замене пиридина дейтеропиридином продуктом гидролиза соли 27и, полученной из трихлорида 2и, оказывается альдегид 29и, дейтерированный по формильной группе.
Следует отметить, что механизм формального переноса гидрид-иона из положения 4 1,4-дигидропиридинового цикла был и в значительной степени остается предметом интенсивных дискуссий, связанных, прежде всего с ключевой ролью такого рода превращений в биохимических реакциях с участием НАДН. В настоящее время механизм, включающий перенос протона и двух электронов, по крайней мере, для биохимических процессов, является общепринятым. С этим механизмом (перенос протона и двух электронов) согласуется и наблюдавшийся нами изотопный обмен водорода при взаимодействии 2,3,4,6-тетраметилбензотрихлорида 2к с дейтеропиридином в присутст-вии гидрохлорида дейтеропиридина C5D5NHCl. Продукт взаимодействия в этих условиях (после гидролиза) на 70 % оказался недейтерированным альдегидом 29к (схема 8).
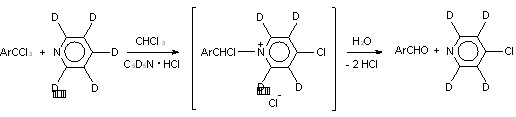 СХЕМА 8
СХЕМА 8
Показано, что в этих условиях дейтерообмен атома водорода -хлорбензильной группы в пиридилпиридиниевых солях типа 27 не происходит.
Таким образом, установлено, что в ходе окислительно-восстановительного процесса водород (дейтерий) переносится на дихлорметиленовую группу N-заместителя не в виде гидрид-иона или атома, а в виде протона (дейтерона), что и определяет высокую вероятность его обмена с другими протонами, присутствующими в реакционной среде. То есть механизм ключевой стадии сопряженного окисления-восстановления однозначно включает перенос протона и два одноэлектронных переноса из положения 4 пиридинового цикла к атому углерода дихлорметиленовой группы N-(,-дихлорбензил)-4-хлор-1,4-дигидропиридина 25.
Таким образом, окислительно-восстановительные превращения о,о’-дизамещенных ТХМА 2и-л под действием пиридиновых оснований 22, очевидно, включают следующие, последовательно протекающие стадии:
- нуклеофильное замещение подвижного атома хлора ТХМА 2и-л пиридиновым основанием по механизму SN1 с образованием хлорида N-(,-дихлорбензил)пиридиния 23;
- нуклеофильное присоединение хлорид-аниона по положению 4 пиридинового цикла соли 23 по механизму SET с образованием N-(,-дихлорбензил)-4-хлор-1,4-дигидропиридина 25;
- перенос протона и двух электронов из положения 4 пиридинового цикла N-(,-дихлорбензил)-4-хлор-1,4-дигидропиридина 25 к атому углерода дихлорметиленовой группы, синхронизированный с отщеплением хлорид-аниона от указанной группы, с ароматизацией пиридинового цикла и образованием N-(-хлорбензил)пиридиниевой соли 28 (в альтернативе – аналогичный, но менее вероятный окислительно-восстановительный процесс превращения из 1-(,-дихлорбензил)-4-(1-пиридинио)-1,4-дигидропиридина 26 в N-(-хлорбензил)пиридил-4-пиридиниевую соль 27);
- нуклеофильное замещение атома хлора в N-(-хлорбензил)пиридиниевой соли 28 пиридиновым основанием с образованием N-(-хлорбензил)пиридил-4-пиридиниевой соли 27 (справедливо только для сильных пиридиновых оснований - 22а-в).
2.3 Природа восстановителя и механизм восстановительной конденсации стерически незатрудненных трихлорметиларенов с гидразинами в пиридине
В разделе 2.2 уже отмечалось, что бензотрихлорид 2а и его метилзамещенные, несущие только одну метильную группу в орто-положении - 2ж,з,м, практически не вступают во взаимодействие с пиридином и его замещенными в среде хлороформа или хлористого метилена при комнатной температуре, однако при кипячении в избытке пиридина образуют биспиридиниевые соли 24ж,з,м с примесью дихлорида 1-(4-пиридил)-пиридиния (выходы 4-6 %). То есть в отличие от высоко реакционноспособных о,о’-диметилзамещенных бензотрихлоридов 2и-л трихлорметиларены 2а,ж,з,м (и вероятно, другие ТХМА с одним орто-заместителем) весьма инертны по отношению к пиридиновым основаниям и способны претерпевать превращения в более жестких условиях преимущественно по пути нуклеофильного замещения двух атомов хлора трихлорметильной группы на остаток пиридина с образованием биспиридиниевых солей 24. Последние, как было нами показано, практически не способны к окислительно-восстановительным превращениям с получением соответствующих ароматических альдегидов 29 (или их производных) и 4-хлор- или 4-пиридилзамещенных пиридинов.
Полученные нами экспериментальные данные и результаты других, последующих работ свидетельствуют о том, что для ТХМА 2а,ж,з,м, характеризующихся невысокой реакционной способностью по сравнению с о,о’-диметилзамещенными ТХМА 2и-л, в условиях восстановительной конденсации может быть реализовано другое, альтернатив-ное изложенному в разделе 2.2 (схема 5) направление окислительно-восстановительных превращений, в котором доминирующим восстановителем выступает не пиридин, а гидразин или гидроксиламин.
Прежде всего, была рассмотрена возможность участия пиридиниевых солей типа 23 и 24 в восстановительной конденсации. Как нами было установлено, при нагревании соли 24а с гидрохлоридом гидразина в пиридине в присутствии воды (в условиях восстановительной конденсации) с высоким выходом получается бензойная кислота, а в отсутствие воды идет гетероциклизация с образованием 3,5-дифенил-1,2,4-триазола. В условиях восстановительной конденсации наблюдался также гидролиз соли 24б, полученной из 2,4-диметилбензотрихлорида 2ж и пиридина.
Для выяснения возможных путей восстановительной конденсации необходимо было исключить превращение о,о’-дизамещенных бензотрихлоридов типа 2и в пиридил-пиридиниевые и хлорпиридиниевые соли 27 и 28, а также другое характерное для трихлоридов типа 2и превращение – нуклеофильное замещение атомов хлора группы ССl3 под действием О- и N-нуклеофилов (воды и гидразинов).
С целью выявления направления восстановительной конденсации, не связанного с промежуточным образованием солей 27 и 28, было изучено взаимодействие мезитотрихлорида 2и с гидразином в присутствии 2-пиколина или 4-пиколина. Эти метилпиридины не образуют с мезитотрихлоридом 4-хлор- или 4-пиридинио-пиридиниевых солей вследствие стерических препятствий. Однако, при этом 4-пиколин дает достаточно стабильную монопиридиниевую соль типа 23. Оказалось, что в обоих случаях наблюдается восстановительная конденсация с образованием 2,4,6-триметил-бензальдазина (13г, Ar = 2,4,6-Ме3С6Н2) (схема 9, путь 1). Взаимодействие мезитотри-хлорида 2и с гидразином в присутствии 2,6-лутидина, неспособного к образованию соответствующих пиридиниевых солей, приводит к продукту неполного восстановления – N-(2,4,6-триметилбензоил)гидразону 2,4,6-триметилбензальдегида 19г (схема 9, путь 2).
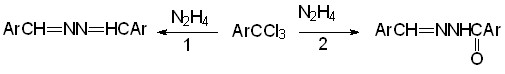
13г 2и 19г
2и, 13г, 19г: Ar = 2,4,6-Me3C6H2
СХЕМА 9
Таким образом, восстановительная конденсация ТХМА с гидразином может проходить без образования пиридиниевых солей 27 и 28, а биспиридиниевые соли 24 в этих условиях не реагируют с гидразином.
На примере восстановления N’-фенилбензогидразоноилхлорида 33а (Ar = R = Ph, R’ = H), было показано, что гидразоноилхлориды 33 способны восстанавливаться под действием гидразина и его замещенных в условиях восстановительной конденсации (0,5 ч кипячения в водном пиридине):
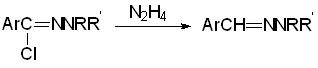
33 34 40 %
СХЕМА 10
В то же время N,N-диметил-2,4,6-триметилгидразоноилхлорид (33б, Аr = 2,4,6-Me3C6H2, R = R’ = Me) и соответствующий ему гидразоноилбромид 33в не изменяются при кипячении с 4-кратным избытком N,N-диметилгидразина в пиридине (схема 11).
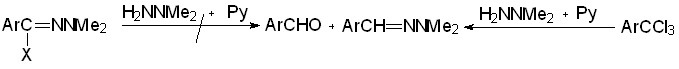
33 29 34 2и
Ar = 2,4,6-Me3C6H2; X = Cl (б), Br (в)
СХЕМА 11
Различие в поведении гидразоноилгалогенидов 33 нельзя объяснить только пространственными препятствиями или недостаточной активностью диметилгидразина как восстановителя. Так, с одной стороны, нами было показано, что мезитотрихлорид 2и при взаимодействии с диметилгидразином в присутствии пиридина дает с суммарным выходом более 80 % смесь 2,4,6-триметилбензальдегида 29и и его N,N-диметилгидразона (34б, Ar = 2,4,6-Me3C6H2, R = R’ = Me; схема 11). С другой стороны, N,N-диметилгидразон 34б был также получен с выходом 37 % при взаимодействии мезитотрихлорида 2и с N,N-диметилгидразином в присутствии 2,6-лутидина.
Еще один возможный интермедиат восстановительной конденсации - N,N-Бис(-хлорбензилиден)гидразин 18а не изменяется при действии пиридина, а в присутствии избытка гидразина и пиридина дает с небольшим выходом бензальдазин 13а (9 %), но основным продуктом взаимодействия оказывается продукт гетероциклизации - 3,6-дифенил-1,2-дигидро-1,2,4,5-тетразин 35 (37 %):
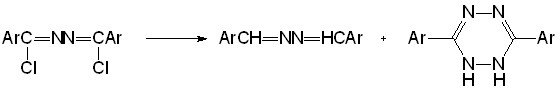
18 13 35 37 %
13а, 18а, 35а, Ar = Ph
СХЕМА 12
Полученные данные не позволяли однозначно считать гидразоноилхлориды 33 интермедиатами восстановительной конденсации. В связи с этим была рассмотрена возможность превращения в гидразоны и азины некоторых соединений, которые могут образоваться из гидразоноилхлоридов в условиях восстановительной конденсации. Учитывая неоднозначность результатов, полученных при попытках восстановления гидразоноилхлоридов, необходимо отметить, что эти опыты проводились в присутствии пиридина и, следовательно, могли возникать гидразоноилпиридиниевые соли типа 36. В использованных условиях в результате взаимодействия гидразоноилхлоридов с гидразинами могли образоваться также гидразидины 37. Наконец, в присутствии основания гидразоноилхлориды (при R’ = H) могли претерпевать дегидрохлорирование, приводящее к нитрилиминам 38.
36 37 38
Поскольку пиридиниевые соли не подвергаются восстановительной конденсации, можно предположить, что и образование солей типа 36 лишь затрудняет или делает невозможным восстановление гидразоноилхлоридов. То, что эта реакция восстановления идет и в отсутствие пиридина, было показано на примере взаимодействия бензотри-хлорида 2а с N,N-диметилгидразином.
Восстановление гидразидина 37 избытком гидразина до гидразона также представляется маловероятным, поскольку эти довольно лабильные соединения очень легко окисляются до формазанов, а восстановление гидразином гидразидинов, в том числе образующихся из гидразоноилхлоридов не наблюдается. Можно полагать, что образование гидразидинов 37, а также тетразина 35 является «тупиковым» процессом, не приводящим к продуктам восстановительной конденсации. В согласии с этим предположением находится и тот обнаруженный факт, что стабильный аналог гидразидинов – N-гидроксимезитоамидоксим – не изменяется в условиях восстановительной конденсации ТХМА с гидроксиламином в пиридине. Что касается возможности образования нитрилиминов 38 в условиях восстановительной конденсации, то реализация этого процесса не подтверждается (см. Беленький Л.И., Луйксаар С.И., Чувылкин Н.Д., Краюшкин М.М. // Изв. АН, Серия хим., 2000, № 5, С.888-895). Известно, в частности, что дифенилнитрилимин (38, R = Ar = Ph) генерируется из N-фенилбензгидразоноилхлорида в присутствии пиридина и присоединяется к последнему по схеме 1,3-диполярного циклоприсоединения с образованием 1,3-дифенил-сим-триазоло[4,3-a]-пиридина, который может быть идентифицирован в виде тетрафторбората 1,3-дифенил-сим-триазоло[4,3-а]-пиридиния. В условиях восстановительной конденсации бензотрихлорида 2а был выделен фенилгидразон бензальдегида (34, Ar = R = Ph, R’ = H), но не удалось обнаружить образования как упомянутого тетрафторбората, так и аддукта дифенилнитрилимина со стиролом. По всей видимости, восстановление гидразоноил-хлорида 33 идет быстрее, чем его дегидрохлорирование с образованием нитрилимина.
Таким образом, второе направление восстановительной конденсации стерически незатрудненных ТХМА 2 с гидразинами в пиридине реализуется с участием гидразинов в качестве восстановителей и образованием,-дихлорбензилгидразина PhCCl2NN’RR’ 39 и гидразоноилхлоридов 33 в качестве интермедиатов.
Резюмируя все изложенное, можно констатировать:
- в условиях восстановительной конденсации ТХМА 2 с гидразинами или гидроксиламином в пиридине могут протекать три конкурирующие направления превращений, два из которых приводят к продуктам восстановления трихлорметильной группы, а третье направление – к продуктам гетероциклизации (1,2,4-оксадиазолам, 1,3,4-оксадиазолам, 1,2,4,5-тетразинам);
- доминирующее направление превращений ТХМА определяется его строением и реакционной способностью;
- наиболее реакционноспособные о,о’-дизамещенные ТХМА 2и-л в условиях восстановительной конденсации или в более мягких условиях (в хлороформе или в хлористом метилене) преимущественно вступают в окислительно-восстановительные превращения с пиридином или его 3-R-замещенными с получением соответствующих ароматических альдегидов 19 или их производных и 4-хлорпиридинов 31 или 1-(4-пиридил)пиридиниевых солей 30; для этих ТХМА процессы восстановления гидразином (гидроксиламином) и гетероциклизации в данных условиях практически не реализуются;
- бензотрихлорид 2а в условиях восстановительной конденсации подвергается превращениям по трём направлениям, из которых основным (доминирующим) является процесс восстановления трихлорметильной группы под действием гидразина (гидроксил-амина) и минорными процессами – гетероциклизация и восстановление под действием пиридина;
- стерически незатрудненные метилзамещенные ТХМА 2 в условиях восстано-вительной конденсации подвергаются превращениям по всем трём направлениям, из которых основными являются процессы восстановления трихлорметильной группы под действием гидразина (гидроксиламина) и пиридина в качестве восстановителей, а процесс гетероциклизации является минорным или вообще не реализуется;
- взаимодействие о,о’-дизамещенных ТХМА 2и-л с пиридином или с его 3-R-замещенными в хлороформе или хлористом метилене представляет собой новый удобный и эффективный метод получения соответствующих ароматических альдегидов и 4-хлор-3-R-пиридинов или 1-(3-R-пиридил-4)-3-R-пиридиниевых солей;
- о,о’-дизамещенные ТХМА 2и-л можно рассматривать в качестве «мягких», -селективных хлорирующих агентов по отношению к пиридину, его 3-R-замещенным и хинолину;
- восстановление трихлорметильной группы ароматических и гетароматических соединений с непосредственным участием пиридиновых оснований в окислительно-восстановительных превращениях носит общий характер и может быть инициировано действием сильных нуклеофилов, причем механизм и направление превращений определяются как строением и реакционной способностью исходного ароматического или гетероароматического соединения, так и природой нуклеофила.
3. Реакции некоторых N- и С-нуклеофилов с солями пиридиния,
полученными из о,о-диметилзамещенных трихлорметиларенов.
3.1. Взаимодействие о,о-диметилзамещенных ТХМА с аминопиридинами
Монопиридиниевые 23, биспиридиниевые 24, а также 1-(4-пиридил)пиридиниевые и 4-хлорпиридиниевые соли 27 и 28 (схема 5) имеют несколько электрофильных центров, которые могут подвергаться нуклеофильной атаке. Так, в рассмотренных в разделе 2.2 превращениях N-(4-пиридил)пиридиниевых или 4-хлорпиридиниевых солей 27 и 28 действие воды или водного этанола приводит к альдегидам 29 и гидрохлоридам 3-R-4-хлорпиридинов 31 или к солям пиридилпиридиния 30, то есть направляется на электрофильный хлорбензильный атом углерода с замещением пиридиниевого остатка и атома хлора на атом кислорода. Аналогичное направление нуклеофильной атаки реализуется и при действии гидразинов или гидроксиламина на соли 27, 28 непосредственно приводя к соответствующим продуктам восстановительной конденсации – гидразонам или оксимам.
Некоторые превращения, не связанные с восстановлением ТХМА, также могут протекать через стадию образования монопиридиниевых солей типа 23 с последующим нуклеофильным замещением одного или двух атомов хлора,-дихлорбензильной группы. Так, это направление реализуется при получении биспиридиниевых солей 24 из стерически незатрудненных ТХМА 2 и пиридина.
Другим превращением указанного типа является взаимодействие о,о-диметил-замещенных ТХМА 2 с 2-аминопиридинами, изученное нами на примере реакций мезитотрихлорида 2и с 2-аминопиридином 22ж или 2-амино-5-бромпиридином 22з при молярном соотношении ТХМА : аминопиридин, равном 1 : 2. Это взаимодействие, осуществляемое в мягких условиях в хлороформе, приводит к необычным амидинам 40а,б с выходами 51-54 % (схема 13).
2и 22 R = H (ж); R = Br (з) 40 R = H (a); R = Br (б)
СХЕМА 13
Строение этих соединений подтверждается данными спектроскопии ЯМР 1Н и 13С, ИК-спектров, а также результатами масс-спектрометрии и элементного анализа.
Образование таких «аномальных» продуктов, вероятно, протекает через стадию образования монопиридиниевой соли типа 23, которая вследствие пониженной электрофильности пиридинового цикла и, в частности, положений 4 и 6, не способна к нуклеофильному присоединению хлорид-аниона и атакуется аминогруппой второй молекулы пиридина с замещением подвижных атомов хлора дихлорметиленового фрагмента. Нуклеофильное замещение атома хлора указанного фрагмента эндоцикли-ческим атомом азота второй молекулы пиридина с образованием биспиридиниевых солей 24, очевидно, невозможно из-за стерических препятствий как со стороны о-метильных групп мезитильного остатка, так и о-аминогруппы пиридинового фрагмента.
Таким образом, на примере реакций мезитотрихлорида 2и с 2-аминопиридинами показано, что взаимодействие реакционноспособных о,о-диметилзамещенных ТХМА 2и-л с пиридинами, имеющими два нуклеофильных центра, может протекать без восстанов-ления трихлорметильной группы.
Однако не все замещенные пиридины, имеющие два нуклеофильных центра, одним из которых является эндоциклический атом азота, способны претерпевать превращения под действием о,о-диметилзамещенных ТХМА по обнаруженному направлению. В связи с этим следует упомянуть, что описанное в разделе 2.2 взаимодействие мезитотрихлорида 2и с 3-гидроксипиридином (22в), тоже имеющим два нуклеофильных центра, приводит к продуктам восстановления трихлорметильной группы – мезитоальдегиду 29и и соответствующей соли 1-(4-пиридил)пиридиния 30в (схема 5).
Очевидно, что направление взаимодействия о,о-диметилзамещенных ТХМА 2и-л с замещенными пиридинами, имеющими два нуклеофильных центра, определяется строением и реакционной способностью исходного пиридинового основания и, в частности, зависит от реакционной способности и расположения каждого нуклеофильного центра.
3.2. Реакции гетарилирования с участием пиридиниевых солей, полученных
из о,о-диметилзамещенных трихлорметиларенов
Одним из центров нуклеофильной атаки пиридиниевых солей 23 или 24 (схемы 4 и 5) является положение 4 пиридинового цикла, что приводит к 4-замещенным пиридинам, в частности, к 4-хлорпиридинам 31 или к солям 1-(4-пиридил)пиридиния 27 и 30. Образование 4-пиридилгидразонов и 4-хинолилгидразона ароматических альдегидов 20б-г и 21 в условиях восстановительной конденсации ТХМА 2ж-и с гидразином также является примером такого направления нуклеофильной атаки. В присутствии сильного нуклеофила – гидразина происходит конкурирующее замещение гидразином (а не пиридином) атома хлора в положении 4 хлорпиридиниевых солей 28, причем образуется 4-пиридилгидразин, который при взаимодействии с соответствующими альдегидами 29 дает 4-пиридилгидразоны 20б-г. Аналогичное объяснение справедливо и для образования 4-хинолилгидразина, превращающегося в соответствующий гидразон 21.
28 29
СХЕМА 14
Образование 4-пиридилгидразина и 4-хинолилгидразина можно рассматривать в качестве первых примеров реакции гетарилирования гидразина, по всей видимости, протекающей с участием соответствующих 4-хлорпиридиниевых (и 4-хлорхинолиниевых) солей 28 или 31.
Одной из задач настоящей работы было выяснение способности пиридиниевых солей 23, 24, 27 и 28 вступать во взаимодействие с различными N- и C-нуклеофилами по схеме реакции гетарилирования, по одному из положений 2 или 4 пиридинового цикла. Решение этой задачи позволило бы выявить и оценить синтетические возможности пиридиниевых солей, генерируемых in situ из доступных исходных реагентов, с целью получения замещенных пиридиновых и хинолиновых оснований.
На примере мезитотрихлорида 2и было показано, что о,о-диметилзамещенные ТХМА 2и-л при взаимодействии с пиридином в хлороформе или хлористом метилене образуют пиридиниевые соли, способные в присутствии пиперидина или морфолина давать продукты гетарилирования. Так, в исследованных нами превращениях, наряду с 2,4,6-триметилбензойным альдегидом 29и, охарактеризованным в виде азина 13г, были получены 4-пиперидинопиридин 41 или 4-морфолинопиридин 42 с выходами соответст-венно 48 % и 57 % (схема 15).
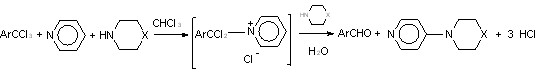
2и--л 41 Х = NН; 42 X = O
СХЕМА 15
Из литературных данных известно, что практически все N-нуклеофилы, в том числе первичные и вторичные алифатические и ароматические амины (за редким исключением), относятся к «жестким» нуклеофилам и, как правило, демонстрируют высокую -селективность присоединения к катионам 1-алкил- и 1-арилпиридиния. Так, ранее было описано присоединение пиперидина к 1-алкил-3-R-замещенным катионам пиридиния, в котором пиперидин выступает как «жесткий» нуклеофил, проявляя исключительно кинетическую -селективность и образуя продукты 1,6-присоединения. Учитывая эти данные, можно констатировать, что исключительная -селективность обнаруженных нами реакций гетарилирования пиперидина и морфолина объясняется тем, что эти реакции протекают последовательно через стадии образования пиридиниевых солей 23 и 28. По всей видимости, именно нуклеофильное замещение пиперидином или морфолином атома хлора в 4-хлорпиридиниевой соли 28и (Ar = 2,4,6-Me3C6H2) ответственно за образование указанных продуктов гетарилирования: 4-пиперидино-пиридина (41) или 4-морфолинопиридина (42) по схеме 16:
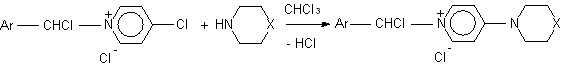
28и
СХЕМА 16
Альтернативным путем получения соединений 41, 42 может быть замещение атома хлора в положении 4 гидрохлорида 4-хлорпиридина 31а соответствующим вторичным амином (схема 17). Однако этот путь представляется менее вероятным, поскольку гидрохлорид 4-хлорпиридина (31а) обладает меньшей реакционной способностью (меньшей электрофильностью) по сравнению с N-замещенной солью 28и.
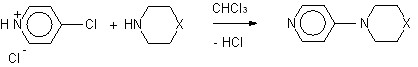
СХЕМА 17
Как уже упоминалось выше, одной из задач настоящей работы являлось выяснение способности пиридиниевых солей типа 23, 24, 27 и 28 вступать во взаимодействие с различными нуклеофилами, в том числе с C-нуклеофилами, по типу реакции гетарилирования. В качестве реагентов для получения наиболее реакционноспособных пиридиниевых солей нами также были выбраны мезитотрихлорид 2и и незамещенный пиридин, а в качестве С-нуклеофилов - -избыточные ароматические системы: N,N-диметиланилин и индол. При этом в мягких условиях были получены соответствующие 4-замещенные пиридины – 4-(4-диметиламинофенил)пиридин 43 и 4-(3-индолил)пиридин 44 с выходами 30 % и 53 % (схемы 18 и 19):
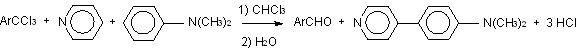
2и 29и 43 30 %
СХЕМА 18
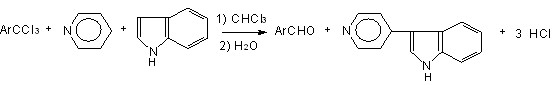
2и 29и 44 53 %
СХЕМА 19
В случае гетарилирования индола наряду с соединением 44 с небольшим выходом (7 %) был выделен 4-(3-индолил)-1-(4-пиридил)-1,4-дигидропиридин 45, получение которого можно рассматривать как первый пример непосредственного присоединения С-нуклеофила к N-замещенной N-(4-пиридил)пиридиниевой соли 27и:
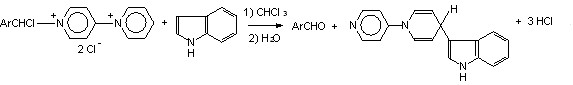
27и 29и 45 7 %
СХЕМА 20
-Селективность этих реакций гетарилирования определяется, по всей видимости, как экранированием -положений пиридиниевой соли объемным N-заместителем –,-дихлор-2,4,6-триметилбензильной группой, так и «мягкой» нуклеофильностью -избыточ-ных систем N,N-диметиланилина и индола, которые атакуют наиболее «мягкий» электрофильный центр С-4 одной из солей пиридиния 23, 28и или 27и.
Касательно образования 4-(3-индолил)-1-(4-пиридил)-1,4-дигидропиридина 45 можно отметить, что аналогичные замещенные 1-(4-пиридил)-1,2- и 1,4-дигидропиридины 46 и 47 были получены при взаимодействии 1-(3-R,5-R-4-пиридил)-3-R,5-R-пириди-ниевых солей 30а,б,г (где R = H, Me; R = H, Me) с трёххлористым фосфором и этанолом (Boduszek B., Wieczorek J.S. // Synthesis, 1979, № 6, p.454).
Рассматривая возможный механизм образования продуктов гетарилирования 41-44 и 45, можно констатировать, что вряд ли он реализуется по типу реакций нуклеофильного замещения N- или C-нуклеофилом остатка пиридиния в 1-(4-пиридил)пиридиниевых солях типа 30а, поскольку подобные превращения протекают в достаточно жестких условиях, отличных от используемых нами «мягких» реакционных условий. С другой стороны, не исключена возможность реакций указанных «жестких» N-нуклеофилов и «мягких» C-нуклеофилов с гидрохлоридом 4-хлорпиридина 31а, образующимся по схеме 6. Как известно, реакции различных нуклеофилов с 4-хлорпиридинами успешно используются для синтеза соответствующих 4-замещенных пиридинов.
Принимая во внимание мягкие условия изученных нами реакций с N- и С-нуклеофилами, трудно однозначно сказать, какая из солей 23, 28 или 31 ответственна за образование продуктов гетарилирования. Отметим при этом, что одними из наиболее изученных и эффективных реагентов гетарилирования являются N-ацилпиридиниевые соли, которые можно рассматривать как близкие аналоги пиридиниевых солей типа 23 и 28, несущие N-заместитель с подобными акцепторными свойствами.
Чтобы проверить способность моно- и биспиридиниевых солей типа 23 и 24 вступать в реакцию гетарилирования, мы попытались получить 4-пиперидино- или 4-морфолинопиридины 41 и 42 с использованием вместо мезитотрихлорида 2и бензотрихлорида 2а, который образует с пиридином биспиридиниевую соль 24а, но не способен в этих условиях к сколько-нибудь заметному образованию 1-(4-пиридил)-пиридиниевых и 4-хлорпиридиниевых солей 27 и 28. Однако эти попытки не привели к успеху: лишь при кипячении реагентов в хлороформе с выходом 4 % был выделен 4-пиперидинопиридин 41. Не удалось также получить желаемые продукты гетарилирования из достаточно стабильной биспиридиниевой соли 24а, предварительно полученной из бензотрихлорида 2а и пиридина. При кипячении в хлороформе соли 24а и пиперидина с последующим добавлением гидразингидрата к реакционной смеси с выходом 7 % был выделен лишь 3,6-дифенил-1,2-дигидро-1,2,4,5-тетразин 35а.
Следовательно, по отношению к указанным «жестким» N-нуклеофилам и «мягким» C-нуклеофилам нестабильные монопиридиниевые соли типа 23 и биспиридиниевые соли 24, получаемые из стерически незатрудненных ТХМА 2а,ж,з и пиридина, недостаточно реакционноспособны для взаимодействия по типу реакций гетарилирования, т.е. по электрофильному положению 2 или 4 пиридиниевого катиона. В то же время монопиридиниевые соли типа 23 и, еще более вероятно, N-замещенные 4-хлорпири-диниевые соли 28, генерируемые из о,о-диметилзамещенных ТХМА 2и-л и пиридина, являются достаточно реакционноспособными и удобными гетарилирующими агентами для N- и C-нуклеофилов. При этом реакции гетарилирования характеризуются исключительно -селективностью по отношению к пиридину. Эта селективность, как можно полагать, определяется как стерическим экранированием -положений пиридиниевой соли 23 объемным N-заместителем -,-дихлор-2,4,6-триметилбензильной группой, так и тем, что гетарилирование «жестких» N-нуклеофилов протекает, по всей видимости, как нуклеофильное замещение атома хлора в положении 4 пиридиниевой соли 28 или гидрохлорида 4-хлорпиридина 31а. В случае же C-нуклеофилов -селективность гетарилирования обусловлена не только стерическим экранированием -положений пиридиниевой соли 23 объемным N-заместителем, но и известным «мягким» характером используемых С-нуклеофилов - -избыточных ароматических соединений.
Таким образом, можно констатировать, что пиридиниевые соли типа 23 и 28, генерируемые из о,о-диметилзамещенных ТХМА 2и-л из пиридина или из 3-R-замещенных пиридинов, являются весьма реакционноспособными и относительно удобными гетарилирующими агентами для различных N- и C-нуклеофилов и могут успешно использоваться для синтеза соответствующих 4-замещенных пиридинов. Принципиальным отличием реакций гетарилирования нуклеофилов с участием пиридиниевых солей 23 и 28, генерируемых из о,о-диметилзамещенных ТХМА 2и-л и пиридина, от реакций гетарилирования с участием других N-замещенных катионов пиридиния является обязательная ароматизация промежуточно возникающих 1,4-дигидропиридинов с одновременным восстановлением,-дихлорбензильной группы N-заместителя.
С учетом известных литературных данных и на основании полученных нами экспериментальных результатов, можно сделать следующие выводы:
- пиридиниевые соли, генерируемых in situ из о,о-диметилзамещенных ТХМА 2и-л и пиридина, весьма близки по своей реакционной способности и свойствам к катионам 1-ацилпиридиния и могут вступать аналогично последним в реакции присоединения (гетарилирования) с различными нуклеофилами;
- о,о-диметилзамещенные ТХМА 2и-л являются удобными алкилирующими агентами, способными эффективно и -селективно активировать пиридиновые основания в их реакциях с «жесткими» и «мягкими» нуклеофилами.
4. Реакции трихлорметиларенов с N-, O- и S-нуклеофилами, протекающие
без восстановления трихлорметильной группы
4.1 Синтезы 2,5-дизамещенных 1,3,4-оксадиазолов на основе
трихлорметиларенов и ацилгидразинов
Ранее при изучении реакции восстановительной конденсации ТХМА с гидразинами и гидроксиламином было обнаружено, что в случае трудно восстанавливающегося бензотрихлорида 2а наблюдается конкуренция между восстановительной конденсацией и гетероциклизацией (раздел 2.1, схема 3).
В связи с этим, одной из задач настоящего исследования было выявление таких условий взаимодействия ТХМА 2 с гидразином или его замещенными, которые были бы оптимальны для селективной гетероциклизации, приводящей к 2,5-дизамещенным 1,3,4-оксадиазолам, и позволили бы полностью исключить или свести к минимуму восстановительную конденсацию с образованием азинов 13 или гидразонов 16.
С этой целью было изучено взаимодействие ТХМА 2 с семикарбазидом и тиосемикарбазидом в условиях восстановительной конденсации и в модифицированных условиях. При взаимодействии ТХМА 2а,ж с семикарбазидом 48а или тиосемикарбазидом 49а в пиридине, помимо соответствующих семикарбазонов и тиосемикарбазонов ароматических альдегидов 50, 51, с выходами до 30 % были выделены 2-амино-5-фенил-1,3,4-оксадиазол 52а и 2-амино-5-арил-1,3,4-тиадиазолы 53а,б.
При этом соотношение двух конкурирующих процессов существенно зависит от строения ТХМА и определяется относительной легкостью восстановления последнего: для бензотрихлорида 2а выходы 1,3,4-оксадиазола 52а и 1,3,4-тиадиазола 53а были выше, чем выходы продуктов восстановительной конденсации 50а и 51а. В случае же 2,4-диметилбензотрихлорида 2ж продукты обеих реакций получались в соотношениях ~ 1:1, а 2,4,6-триметилбензотрихлорид 2и давал только соответствующие продукты восстановле-ния – семикарбазон 50в и тиосемикарбазон 51в.
Изменение условий проведения процесса, и в частности использование в качестве растворителя смеси пиридин – метанол, в общем случае привело к увеличению выходов продуктов гетероциклизации – 52а и 53а,б. Так, при взаимодействии бензотрихлорида 2а с семикарбазидом 48а или тиосемикарбазидом 49а в метанольно-пиридиновой смеси единственными продуктами превращений оказались соответственно 2-амино-5-фенил-1,3,4-оксадиазол 52а и 2-амино-5-фенил-1,3,4-тиадиазол 53а с выходами 63 % и 60 % (схема 21).
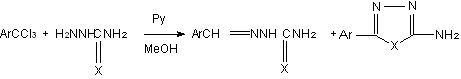
2а,ж,и 48а Х = О 50 X = O 52а: Ar = Ph, X = O (63 %)
49а Х = S 51 X = S 53: а Ar = Ph; б Ar = 2,4-Me2C6H3
СХЕМА 21
Таким образом, было показано, что использование в качестве растворителя смеси пиридина и метанола позволяет свести к минимуму процесс восстановительной конденсации (за исключением высоко реакционноспособного мезитотрихлорида 2и) и существенно повысить выход продуктов гетероциклизации – 2-амино-5-арил-1,3,4-оксадиазолов (52) или 1,3,4-тиадиазолов (53).
С целью оптимизации условий гетероциклизации и выявления зависимости выхода целевых продуктов от строения и реакционной способности реагентов, мы изучили особенности взаимодействия шести ТХМА (2а,ж-и,м,н), отличающихся наличием и характером заместителей, и двенадцати гидразидов: семикарбазида (48а), производных алифатических (48б), ароматических (48в-з) и гетероциклических (48и-н) карбоновых кислот в различных условиях, отличных от условий восстановительной конденсации. Общая схема исследуемых превращений ТХМА 2 с целью получения 2,5-дизамещенных 1,3,4-оксадиазолов приведена ниже (схема 22).
Логичным способом оптимизации условий гетероциклизации мог бы быть отказ от использования в качестве растворителя пиридина, поскольку последний, как показано нами выше, участвует в самом акте восстановления. Однако первые попытки исключить участие пиридина в исследуемой реакции не привели к успеху.
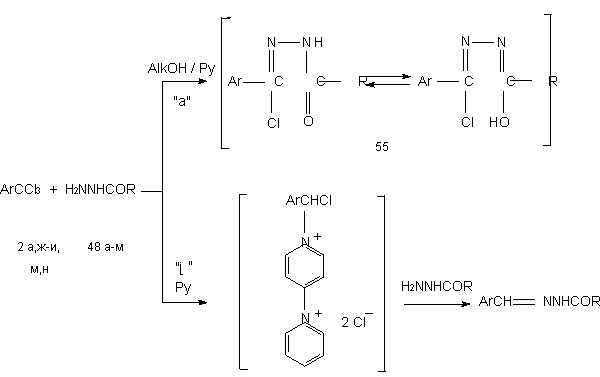
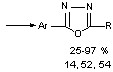
27 16, 19, 50
СХЕМА 22
Так, в спиртовом растворе в присутствии карбоната натрия как основания при использовании метанола или этанола в качестве растворителей с переходом от бензотрихлорида 2а к его метилзамещенным 2ж и 2з,и резко ускоряется алкоголиз трихлорметиларенов. При этом основными продуктами реакции становятся сложные эфиры соответствующих замещенных бензойных кислот, а выходы оксадиазолов 14, 52, 54 не превышают 25 %. В случае же мезитотрихлорида 2и эфиры 2,4,6-триметил-бензойной кислоты оказались единственными продуктами реакции с выходом до 88 %.
Аналогичные результаты были получены при использовании морфолина или пиперидина в качестве оснований при взаимодействии 2,4,5-триметилбензотрихлорида 2з с 2-гидроксибензгидразидом 48г в метанольном растворе: выходы 2,5-диарил-1,3,4-оксадиазола 14н колебались в пределах 12-22 %, причем основным продуктом реакции оказался метиловый эфир 2,4,5-триметилбензойной кислоты (выходы 56 % и 63 %).
Применение же в качестве растворителя и основания триэтиламина, хотя и исключает возможность алкоголиза, не приводит к увеличению выхода оксадиазолов. При взаимодействии в метаноле или этаноле в отсутствие основания преимущественно протекает алкоголиз трихлорметиларенов.
Из апробированных условий наиболее оптимальные результаты даёт кипячение реагентов в течение 6-15 часов в метанольно-пиридиновом или этанольно-пиридиновом растворе при объемном соотношении спирт : пиридин от 2:1 до 5:1. В этих условиях выходы целевых 1,3,4-оксадиазолов находятся в пределах 22-97 % и заметно превышают выходы, достигаемые в других условиях. Добавление карбоната натрия к спиртово-пиридиновой смеси приводит лишь к снижению выхода 1,3,4-оксадиазолов 14 и 54 вследствие более эффективного алкоголиза ТХМА, промотируемого неорганическим основанием.
Рассматривая влияние природы исходных реагентов на выходы 1,3,4-оксадиазолов 14, 52 и 54, можно констатировать, что эффект строения ароматических гидразидов 48 проявляется довольно умеренно. Для реакций ТХМА 2а,ж-и,м с гидразидами кислот гетероароматического ряда 48и-м характерно некоторое снижение выхода целевых продуктов 54а-о по сравнению с выходами оксадиазолов 14 в реакциях ТХМА с гидразидами ароматических кислот 48в-з, причем особенно отчётливо это снижение проявляется для гидразидов 48и,к – производных 4,5-дибром-2-фуранкарбоновой и 2-тиофенкарбоновой кислот.
При переходе от незамещенного бензотрихлорида 2а к метилзамещенным ТХМА 2ж,з,и выходы 1,3,4-оксадиазолов в одинаковых условиях, как правило, снижаются с увеличением числа метильных групп в ароматическом кольце, что объясняется ускорением в том же гомологическом ряду параллельно протекающего алкоголиза ТХМА. В случае превращений наиболее реакционноспособного и легко восстанавливающегося 2,4,6-триметилбензотрихлорида 2и даже в оптимальных для гетероциклизации условиях, то есть при нагревании ТХМА и гидразида в спиртово-пиридиновом растворе, целевые 1,3,4-оксадиазолы получить не удается. Причем в этих условиях получаются лишь продукты двух конкурирующих процессов: замещенные гидразоны мезитоальдегида (19, 50), образующиеся в результате восстановительной конденсации, и эфиры 2,4,6-три-метилбензойной кислоты, получающиеся в результате алкоголиза трихлорида 2и.
С целью выяснения механизма взаимодействия бензотрихлоридов 2 с гидразидами 48 в спиртово-пиридиновом растворе и, в частности, выяснения роли пиридина, мы проверили возможность участия в гетероциклизации моно- (23) и биспиридиниевых солей (24), возникающих при взаимодействии трихлорметиларенов с пиридином по схеме 5. Нами было показано, что предварительно полученная биспиридиниевая соль 24з (Ar = 2,4,5-Me3C6H2) способна при взаимодействии с бензгидразидом 48в в метаноле превращаться в оксадиазол 14м (выход составил 14 %), однако, обнаружить образование солей 24 в указанных условиях гетероциклизации так и не удалось. С другой стороны, в условиях гетероциклизации могут образовываться N,N’-диароилгидразины 17 или N-ароил-N-гетароилгидразины, например, в результате алкоголиза трихлорметиларенов. В частности, при взаимодействии 2,4,5-триметилбензотрихлорида 2з с салицилгидразидом 48г в метанольно-пиридиновом растворе наряду с оксадиазолом 14н в незначительном количестве был выделен N-(2,4,5-триметилбензоил)-N’-(2-гидроксибензоил)гидразин 17б. Однако, на примере предварительно полученного нами N-бензоил-N’-(3-нитробензоил)-гидразина 17в было показано, что такие N,N’-диацилгидразины (17) не циклизуются при кипячении в метанольно-пиридиновом растворе. Наиболее вероятным представляется участие в гетероциклизации промежуточно возникающих гидразоноилхлоридов 55 или сложных эфиров N-ацилбензгидразоновых кислот типа 56, которые могут получаться при алкоголизе гидразоноилхлоридов 55. Реальность такой последовательности процесса гетероциклизации подтверждается выделением из реакционной смеси небольшого количества этилового эфира N’-ацетилбензгидразоновой кислоты 56а (выход 12 %), образование которого показано ниже (схема 23):
 12 %
12 %
55а Ar = Ph, R = CH3 56а Ar = Ph, R = CH3
СХЕМА 23
При использовании метанола соответствующий метиловый эфир N-ацетилбенз-гидразоновой кислоты выделить не удалось, что согласуется с литературными данными о существенно большей лёгкости циклизации в 1,3,4-оксадиазолы метиловых эфиров N-ацилбензгидразоновых кислот по сравнению с этиловыми эфирами типа 56.
Состав смесей и строение синтезированных соединений подтверждается данными элементного анализа, масс-спектрометрии, ИК- и ЯМР-спектроскопии. В ИК-спектрах всех 1,3,4-оксадиазолов имеются полосы поглощения в области 1620-1600 см-1 (С=N) и 1190-1100 см-1 (С-О-С), которые характерны для оксадиазольного цикла и согласуются с имеющимися литературными данными.
Таким образом, был разработан простой одностадийный метод синтеза 2,5-дизамещенных 1,3,4-оксадиазолов на основе ТХМА 2 и ацилгидразинов, обозначена область применения этого метода, включая возможность использования гидразидов гетероароматических кислот, изучена зависимость выхода оксадиазолов от строения исходных соединений и получены данные о механизме гетероциклизации. Последний, по всей видимости, включает промежуточное образование и циклизацию гидразоноил-хлоридов 55 и эфиров N’-ацилбензгидразоновых кислот 56.
4.2. Синтез симметрично замещенных 2,5-диарил-1,3,4-оксадиазолов
взаимодействием трихлорметиларенов с гидразингидратом
Как было показано выше, при изучении реакций трихлорметиларенов 2 с ацилгидразинами 48 были выявлены основные факторы, определяющие направление превращений. В частности, в спиртовых растворах в отсутствие пиридина из бензотрихлорида 2а и его метилзамещенных 2ж,з,и в результате алкоголиза получаются, главным образом, эфиры ароматических карбоновых кислот, тогда как 2,5-дизамещенные 1,3,4-оксадиазолы 14, 52 и 54 образуются в качестве минорных продуктов. При взаимодействии этих же реагентов в пиридиновых растворах преимущественно или исключительно получаются продукты восстановительной конденсации – соответствую-щие N–замещенные гидразоны ароматических альдегидов 50. Оптимальным для гетероциклизации, приводящей к замещенным 1,3,4-оксадиазолам 14, 52 и 54, оказалось кипячение реагентов в смеси пиридина с метанолом или этанолом:
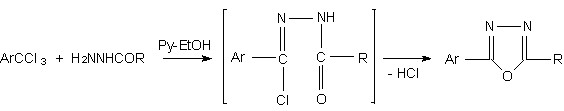
2 48 55 14, 52, 54 22-97 %
СХЕМА 24
Вместе с тем, при кипячении ТМХА 2а,ж с гидразингидратом N2H4H2O (мольное соотношение ТХМА : гидразингидрат равно 2 : 1) в смеси пиридин-метанол симметрично замещенные 2,5-диарил-1,3,4-оксадиазолы 14а,у были получены с довольно низкими выходами - 17 % и 25 %. При этом в случае бензотрихлорида (2а) наряду с 2,5-дифенил-1,3,4-оксадиазолом (14а) были выделены метилбензоат (выход 43 %) и бензгидразид 48в (выход 8 %).
Учитывая эти результаты, с целью более эффективного синтеза симметрично замещенных 1,3,4-оксадиазолов было решено отказаться от использования пиридина в качестве сорастворителя и акцептора хлористого водорода и проводить реакцию в иных условиях – при кипячении в этаноле в присутствии избытка гидразингидрата в качестве акцептора НСl. Было показано, что кипячение реагентов в этаноле в течение 40 минут приводит к 2,5-дифенил-1,3,4-оксадиазолу (14а, Ar = R) с выходом 96 %:
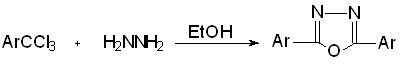
2а,в,н 14а,ф,х 68-96 %
2: Ar = Ph (а); 4-ClC6H4 (в); 3-BrC6H4 (н); 14: Ar = Ph (а); 4-ClC6H4 (ф); 3-BrC6H4 (х)
СХЕМА 25
В указанных условиях аналогично бензотрихлориду 2а с гидразином реагируют галогензамещенные ТХМА 2в,н, проявляющие более низкую реакционную способность, – 4-хлорбензотрихлорид (2в) и 3-бромбензотрихлорид (2н), при этом выходы оксадиазолов 14ф,х составляют соответственно 81 % и 68 %. В случае о-замещенного ТХМА, 2-хлор-бензотрихлорида 2б, был выделен только продукт последовательного гидролиза и алкоголиза – этиловый эфир 2-хлорбензойной кислоты (78 %). Очевидно, в данных условиях вследствие стерических препятствий, обусловленных о-заместителем 2-хлор-бензотрихлорида 2б, конкурирующий алкоголиз трихлорметильной группы протекает намного быстрее, чем взаимодействие ТХМА 2б с гидразингидратом с промежуточным образованием гидразоноилхлорида 55, а возникающий при алкоголизе этиловый эфир 2-хлорбензойной кислоты вследствие тех же стерических препятствий и в условиях кратковременного взаимодействия практически не подвергается гидразинолизу.
В тех же условиях высоко реакционноспособный мезитотрихлорид 2и, который в спиртово-пиридиновых смесях дает лишь продукты восстановительной конденсации и алкоголиза, также не удалось подвергнуть гетероциклизации, так как он полностью превращается в этиловый эфир 2,4,6-триметилбензойной кислоты (выход 95 %). Это свидетельствует о том, что в спиртовой среде для высоко реакционноспособных о,о’-дизамещенных ТХМА 2 доминирующим процессом является алкоголиз.
С учетом полученных нами результатов детальная схема синтеза симметрично замещенных 1,3,4-оксадиазолов для реакций стерически незатрудненных ТХМА 2 с гидразингидратом в спиртовой среде включает в себя следующие стадии (схема 26):
- алкоголиз ТХМА с образованием дихлорацеталя 57, либо неполный гидролиз под действием воды с образованием ароилхлорида 58, либо образование гидразоноилхлорида 33 под действием гидразина (параллельно протекающие реакции);
- превращение дихлорацеталя 57 в ароилхлорид 58 с отщеплением RCl, либо под действием гидразина в эфир гидразинокислоты 59; альтернативный путь образования эфира гидразинокислоты 59 – взаимодействие гидразоноилхлорида 33 со спиртом (алкоголиз);
- взаимодействие эфира гидразинокислоты 59 с ароилхлоридом 58 или с дихлор-ацеталем 57 с образованием эфира N-ацилбензгидразоновой кислоты 56;
- циклизация эфира N-ацилбензгидразоновой кислоты 56 с отщеплением спирта в симметрично замещенный 1,3,4-оксадиазол; альтернативный путь – циклизация под действием основания гидразоноилхлорида 55, образующегося при взаимодействии ароилхлорида 58 или дихлорацеталя 57 с гидразоноилхлоридом 33.
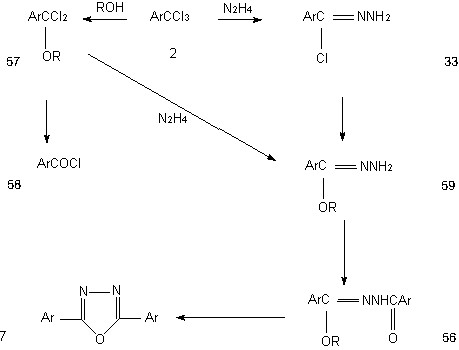
СХЕМА 26
Нежелательным, конкурирующим направлением превращений ТХМА 2 является взаимодействие ароилхлоридов 58 со спиртом с образованием эфиров соответствующих бензойных кислот, которые в условиях кратковременного взаимодействия практически не подвергаются гидразинолизу с образованием гидразидов кислот. Это направление взаимодействия преимущественно реализуется для стерически затрудненных ТХМА 2б,и. Так, 2-хлорбензотрихлорид 2б и мезитотрихлорид 2и, по всей видимости, реагируют не с молекулами спирта или гидразина, а с молекулами воды (из гидразингидрата), образуя ароилхлориды 58, которые легко превращаются в сложные эфиры соответствующих ароматических кислот. Последние, как мы показали экспериментально, в данных условиях не образуют в сколько-нибудь заметных количествах гидразиды ароматических кислот.
Таким образом, полученные нами результаты позволяют констатировать, что хорошие препаративные выходы симметричных 2,5-диарил-1,3,4-оксадиазолов 14 при взаимодействии ТХМА с гидразингидратом в этаноле (метаноле) можно достичь лишь для стерически незатрудненных ТХМА 2а,в,н, проявляющих к тому же невысокую реакционную способность. Для других ТХМА 2, в том числе для всех метилзамещенных гомологов бензотрихлорида 2ж-м, в этих условиях доминирующим процессом является алкоголиз с образованием сложных эфиров бензойных кислот.
4.3. Синтезы 1,4-бис-(5-R-1,3,4-оксадиазолил-2)бензолов
С целью получения полиядерных гетероциклических соединений, обладающих люминесцентными свойствами, мы распространили разработанный нами метод получения 2,5-диарилзамещенных 1,3,4-оксадиазолов 14 и 54 (см. раздел 4.1) на бифункциональный, выпускаемый в промышленном масштабе ТХМА - 1,4-бис(трихлорметил)бензол 2д, технология получения которого была нами разработана и запатентована.
Взаимодействие ТХМА 2д с некоторыми ацилгидразинами (48в,г,ж,м,н) в этанольно-пиридиновой смеси позволило нам получить ряд ранее описанных 1,4-бис-(5-R-1,3,4-оксадиазолил-2)бензолов 60в,г,ж,м,н с умеренными выходами в пределах 35-47 %. Обсуждаемые превращения представлены ниже на схеме 27:
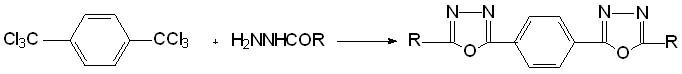
2д 48 60 35-47 %
48, 60: R = Ph (в); 2-OHC6H4 (г); 4-NO2C6H4 (ж), 4-C5H4N (м); H (н);
СХЕМА 27
Синтез 1,4-фениленбис-1,3,4-оксадиазолов типа 60 на основе бис(трихлорметил)-аренов и гидразидов кислот 48 до настоящей работы не был описан, хотя многие подобные гетероциклические системы, особенно с заместителем R = Ar, обладающие люминесцентной способностью, хорошо известны. Описанные ранее методы получения этих полиядерных гетероциклов характеризуются многостадийностью и основаны на относительно сложном препаративном синтезе исходных соединений, что весьма существенно ограничивает препаративную ценность и область применения таких методов синтеза, а также обусловливает высокую производственную себестоимость целевых продуктов.
Несмотря на то, что выходы 1,4-фениленбис-1,3,4-оксадиазолов 60в,г,ж,м,н по разработанному нами методу относительно невысоки (как и в случае диарилоксадиазолов 14, 52 и 54, это обусловлено параллельно протекающим алкоголизом), его несомненными преимуществами являются: доступность исходных соединений и растворителей; универсальность и простота синтеза (в одну стадию); возможность его реализации в промышленном масштабе с использованием обычного технологического оборудования.
Фениленбис-1,3,4-оксадиазолы 60 обладают весьма низкой растворимостью в большинстве обычных растворителей, что ограничивает возможность использования для их анализа и идентификации ЯМР-спектроскопии. Поэтому для доказательства строения синтезированных соединений нами получены и детально рассмотрены их масс-спектры электронного удара. При этом были впервые выявлены специфические направления фрагментации 1,4-фениленбис-1,3,4-оксадиазолов, пригодные для чёткой идентификации и подтверждения строения этих гетероциклических систем.
4.4. Особенности взаимодействия о,о’-дизамещенных трихлорметиларенов с ацилгидразидами при гетероциклизации
Как уже отмечалось, в реакциях трихлорметиларенов с гидразином и его производ-ными в пиридине или в смеси пиридина с метанолом (этанолом) соотношение двух конкурирующих процессов – восстановления ТХМА и гетероциклизации – зависит, в первую очередь, от строения исходного ТХМА 2 и определяется относительной легкостью восстановления последнего. Причем в присутствии спирта протекает и третий конкуриру-ющий процесс – алкоголиз трихлорметильной группы. Дальнейшее изучение взаимодей-ствия ТХМА 2 с гидразином или ацилгидразинами показало, что проведение реакции в смесях пиридина с этанолом или метанолом позволяет существенно увеличить (до 97 %) выходы продуктов гетероциклизации – 2,5-дизамещенных 1,3,4-оксадиазолов 14, 52 и 54. Однако, даже в этих, оптимальных для гетероциклизации условиях при взаимодействии мезитотрихлорида (2и) с гидразином и гидразидами карбоновых кислот (48) получить целевые мезитилзамещенные 1,3,4-оксадиазолы не удалось. В этом случае получались лишь продукты восстановительной конденсации – арилгидразоны 2,4,6-триметил-бензальдегида 19 и/или продукты алкоголиза - эфиры 2,4,6-триметилбензойной кислоты, причем выход последних в отсутствие пиридина достигал 88-95 %.
Такое специфическое поведение мезитотрихлорида, несомненно, обусловлено его строением и связанной с ним высокой реакционной способностью. Согласованный эффект трех метильных групп резко облегчает любое нуклеофильное замещение атомов хлора группы ССl3. В итоге направление реакции определятся конкуренцией присутствующих в реакционной смеси трёх нуклеофилов: гидразида, пиридинового основания и спирта. Последние два, как показали полученные нами результаты, оказываются наиболее сильными. В связи с этим представлялось весьма актуальным, как с теоретической, так и с практической точки зрения, найти такие условия реакции, которые позволили бы селективно получить 2,5-диарил-1,3,4-оксадиазолы из стерически затрудненных о,о’-дизамещенных трихлорметиларенов 2и-л. Основная задача при этом состояла в необходимости заблокировать нежелательные процессы алкоголиза и восстановительной конденсации путем использования других, более инертных растворителей и оснований. Так, восстановительную конденсацию можно предотвратить, если использовать в качестве основания и сорастворителя - или -метилпиридин, неспособный участвовать в акте восстановления.
Эта идея была успешно реализована в работе (Беленький Л.И., Луйксаар С.И., Краюшкин М.М., Химия гетероцикл. соедин., 1999, № 4, С.557-563) при использовании трет-бутанола вместо первичных спиртов (этанола, метанола), что позволило подавить нежелательный алкоголиз. При этом в качестве основания вместо пиридина был использован 2,6-диметилпиридин, который, как было показано нами ранее, неспособен из-за стерических препятствий образовывать с мезитотрихлоридом 2и пиридиниевые соли 23 и восстанавливать трихлорметильную группу. В итоге из высоко реакционноспособного мезитотрихлорида 2и и гидразидов 48в,г,е,о были получены 5-арил(гетарил)-2-(2,4,6-триметилфенил)-1,3,4-оксадиазолы 14ц-ш и 54п с выходами 50-80 %:
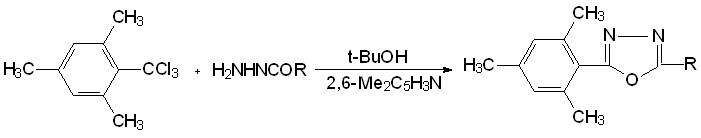
2и 48 14, 54 50-80 %
48: в R = Ph; г R = 2-НOC6H4; е R = 3-O2NC6H4; о R = 2-C4H3O (фурил).
14: ц R = Ph; ч R = 2-НOC6H4; ш R = 3-O2NC6H4; 54п R = 2-C4H3O (фурил).
СХЕМА 28
Таким образом, были найдены такие условия, которые обусловливают селективное взаимодействие о,о’-дизамещенных трихлорметиларенов с ацилгидразинами по реакции гетероциклизации с образованием дизамещенных 1,3,4-оксадиазолов. Можно констати-ровать, что данные условия являются необходимыми и достаточными для селективной гетероциклизации любых ТХМА 2 под действием ацилгидразинов или гидразингидрата, причем в качестве основания может быть использован более доступный 4-метилпиридин, который не вступает в реакцию восстановительной конденсации с ТХМА.
4.5 Синтез 2,5-дизамещенных 1,3,4-тиадиазолов взаимодействием
трихлорметиларенов с тиоацилгидразинами
Как было показано в разделе 4.1, взаимодействие ТХМА 2а,ж с тиосемикарбазидом 49а в метанольно-пиридиновой смеси позволяет получать продукты гетероциклизации – 2-амино-5-арил-1,3,4-тиадиазолы 53а,б с минорными количествами продуктов восстано-вительной конденсации – тиосемикарбазонов. Было естественно предположить, что найденные нами условия гетероциклизации с образованием 2-амино-5-арил-1,3,4-тиадиазолов или 2,5-дизамещенных 1,3,4-оксадиазолов могут оказаться подходящими для синтеза 2,5-дизамещенных 1,3,4-тиадиазолов на основе ТХМА 2 и соответствующих тиоацилгидразинов 49.
И действительно, взаимодействие ТХМА 2а,ж с тиобензгидразидом 49б при кипячении в метанольно-пиридиновой смеси привело к получению 2-арил-5-фенил-1,3,4-тиадиазолов 53в,г с выходами 65 % и 50 % соответственно. Эти результаты являются первыми примерами простого, одностадийного синтеза 2,5-диарил-1,3,4-тиадиазолов:
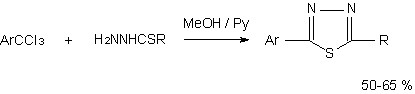
2а,ж 49б: R = Ph 53: Ar = Ph (в); Ar = 2,4-Me2C6H3 (г)
СХЕМА 32
Сами ТХМА 2 могут выступать как удобные исходные соединения для получения тиоацилгидразинов 49, что и было экспериментально продемонстрировано на примере синтеза тиобензгидразида 49 из бензотрихлорида 2а, гидросульфида натрия и гидразин-гидрата. Выход тиобензгидразида составил 70 %.
* * *
Таким образом, проведенные исследования позволили определить оптимальные условия селективного одностадийного синтеза разнообразных 2,5-дизамещенных 1,3,4-оксадиазолов и 1,3,4-тиадиазолов из доступных трихлорметиларенов и гидразина или ацил(тиоацил)гидразинов, выявить особенности и основные закономерности процесса гетероциклизации в зависимости от строения и реакционной способности ТХМА и гидразинов. На основании полученных результатов можно констатировать следующее:
- кипячение ТХМА и ацил- или тиоацилгидразинов в спиртовой среде в присутст-вии пиридинового основания в качестве акцептора хлористого водорода представляет собой оптимальные условия процесса гетероциклизации;
- пиридин и его метилзамещенные (4-метилпиридин, 2,6-диметилпиридин и 2,4,6-триметилпиридин) являются наиболее эффективными основаниями, оказывающими непосредственное влияние на селективную гетероциклизацию ТХМА и ацилгидразинов, причем эффект пиридинового основания, по всей видимости, не ограничивается только ролью акцептора хлористого водорода, но и включает активирующее действие основания на ТХМА в реакциях с ацилгидразином и на циклизацию промежуточно возникающих гидразоноилхлоридов;
- в одинаковых условиях реакции выход целевых продуктов гетероциклизации определяется, в первую очередь, строением и реакционной способностью ТХМА и, в меньшей степени, зависит от природы ацилгидразинов;
- использование третичного спирта (трет-бутанола) в качестве растворителя и 4-метилпиридина, 2,6-диметилпиридина или 2,4,6-триметилпиридина в качестве основания позволяет минимизировать или полностью исключить нежелательные процессы алкоголиза и восстановительной конденсации и обеспечить селективное получение 2,5-дизамещенных 1,3,4-оксадиазолов и 1,3,4-тиадиазолов из любых трихлорметиларенов и ацилгидразинов или гидразингидрата.
Выводы
1. Установлены основные закономерности селективного радикального хлорирова-ния метилбензолов с получением трихлорметиларенов, бензилидендихлоридов и бензил-хлоридов. Систематически изучены основные направления реакций трихлорметиларенов с N-, О- и S-нуклеофилами – гетероциклизации с получением 2,5-дизамещенных 1,3,4-оксадиазолов или 1,3,4-тиадиазолов, гидролиза, ацидолиза или алкоголиза с получением хлорангидридов или сложных эфиров бензойных кислот и восстановительной конденса-ции трихлорметиларенов с гидразинами в пиридине, приводящей к производным соответствующих ароматических альдегидов. Впервые найдены условия, оптимальные для селективной реализации одного из возможных, конкурирующих процессов взаимодействия трихлорметиларенов с N-, О- и S-нуклеофилами.
2. Разработан общий, промышленно значимый способ радикального галогениро-вания алкилароматических, алифатических насыщенных и линейных непредельных углеводородов с использованием высокоэффективных стабилизаторов – эфиров ортофосфорной кислоты, образующих стабильные, недиссоциирующие комплексы с кислотами Льюиса и дезактивирующих каталитический эффект в побочных реакциях электрофильного замещения и дегидрогалогенирования.
3. Впервые определены условия селективного радикального хлорирования метилбензолов в присутствии эфиров ортофосфорной кислоты с получением продуктов заданной степени хлорирования с высокими выходами, высокой конверсией хлора и высокой производительностью процесса.
4. Систематически исследован механизм восстановительной конденсации трихлор-метиларенов с гидразинами в пиридине и впервые установлено, что восстановителем является либо пиридин, либо гидразин в зависимости от наличия и природы заместителей в орто-положениях трихлорметиларена.
5. Восстановительная конденсация трихлорметиларенов, не имеющих орто-заместителей или имеющих один орто-заместитель, преимущественно осуществляется при действии избытка гидразина на первоначально образующиеся гидразоноилхлориды без участия пиридина в акте восстановления. Показана принципиальная возможность реализации этого направления восстановительной конденсации для трихлорметиларенов, имеющих два орто-заместителя.
6. Окислительно-восстановительные превращения о,о-дизамещенных трихлор-метиларенов, протекающие через стадию образования солей N-(,-дихлорбензил)-пиридиния, являются общими в ряду пиридиновых оснований и характерны для 3-R-замещенных пиридинов и хинолина. В зависимости от нуклеофильности атома азота гетероцикла взаимодействие с о,о-дизамещенными ТХМА приводит либо к N-(-хлорбензил)-4-хлор-3-R-пиридиниевым, либо к N-[N-(-хлорбензил)-3-R-4-пиридил]-3-R-пиридиниевым солям. Обнаруженные и изученные превращения моделируют окислительно-восстановительные биохимические процессы с участием никотинамид-адениндинуклеотида (НАДН) и его фосфата и представляют собой новые эффективные методы синтеза о,о-дизамещенных ароматических альдегидов или их производных, а также 4-хлорпиридинов и N-(4-пиридил)пиридиниевых солей.
7. Пиридиниевые соли, генерируемые in situ из о,о-дизамещенных трихлорметил-аренов и пиридинов, являются реакционноспособными гетарилирующими агентами для N- и С-нуклеофилов. Гетарилирование с участием пиридиниевых солей характеризуется исключительной -селективностью по отношению к пиридину независимо от «жесткого» или «мягкого» характера используемых нуклеофилов. Впервые показана возможность селективного -гетарилирования С-нуклеофилов 1-(4-пиридил)пиридиниевыми солями с образованием N-(4-пиридил)-4-R-замещенных 1,4-дигидропиридинов.
8. о,о-Диметилзамещенные трихлорметиларены являются удобными алкилирую-щими агентами для -селективной активации пиридиновых оснований в реакциях гетарилирования как «жестких», так и «мягких» нуклеофилов.
9. Впервые найдены оптимальные условия для селективной гетероциклизации трихлорметиларенов под действием N-, О-нуклеофилов. Разработаны простые и эффек-тивные методы синтеза 2,5-дизамещенных 1,3,4-оксадиазолов, 1,4-фениленбис-1,3,4-оксадиазолов из трихлорметиларенов и гидразидов карбоновых кислот или гидразин-гидрата. Показано, что пиридин и его 2-метил- и 4-метилзамещенные являются наиболее эффективными основаниями, которые активируют исходные трихлорметиларены и обусловливают селективное и быстрое протекание гетероциклизации.
10. Разработан новый, одностадийный препаративный метод синтеза 2,5-дизаме-щенных 1,3,4-тиадиазолов на основе трихлорметиларенов и тиоацилгидразинов.
11. Строение всех синтезированных соединений и интермедиатов изученных превращений трихлорметиларенов установлено методами спектроскопии ЯМР 1Н, 13С и масс-спектрометрии. Выявлены специфические направления фрагментации 2,5-диарил-1,3,4-оксадиазолов и 1,4-фениленбис-1,3,4-оксадиазолов, пригодные для чёткой идентификации и подтверждения строения этих гетероциклических систем.
12. Существенно расширен синтетический потенциал трихлорметиларенов в промышленном и препаративном синтезе соединений и химических продуктов: аромати-ческих альдегидов и их производных, 4-замещенных пиридинов, 2,5-дизамещенных 1,3,4-оксадиазолов и 1,3,4-тиадиазолов, многофункциональных модификаторов резиновых смесей и полимерной серы.
Основное содержание работы изложено в следующих работах:
Обзоры и статьи в журналах, рекомендованных ВАК
1. Поддубный И.С. Региоселективность реакций пиридиниевых и хинолиниевых солей с различными нуклеофилами. // Химия гетероцикл. соедин.- 1995. - № 6.- С.774-815.
2. Беленький Л.И., Поддубный И.С., Луйксаар С.И., Краюшкин М.М. Трихлор-метиларены в синтезе 1,3,4-оксадиазолов. // В Кн. «Химия и биологическая активность синтетических и природных соединений. Азотистые гетероциклы и алкалоиды». / Под. ред. В.Г.Карцева и Г.А.Толстикова. М.: Иридиум-пресс, 2001. - Т.1. - С.46-52.
3. Беленький Л.И., Поддубный И.С., Краюшкин М.М. О начальных стадиях восстановительной конденсации трихлорметиларенов с гидроксиламином и гидразинами в пиридине. // Изв. АН, Серия химическая. - 1993. - № 11. - С.1928-1931.
4. Belen’kii L.I., Poddubny I.S., Krayushkin M.M. Reaction of Trichloromethylarenes with Pyridine: A Novel Synthesis of N-(4-Pyridyl)pyridinium Salts and Aromatic Aldehydes. // Mendeleev Communication. - 1993. - Р.97, 98.
5. Belen’kii L.I., Poddubny I.S., Krayushkin M.M. Synthesis of 2-Amino-5-aryl-1,3,4-thiadiazoles from Trichloromethylarenes: The Effect of Reaction Conditions. // Phosphorus, Sulfur and Silicon, 1994, Vol. 95-96, Р.469-470.
6. Поддубный И.С., Беленький Л.И., Краюшкин М.М. Синтез 2,5-дизамещенных 1,3,4-оксадиазолов на основе трихлорметиларенов и ацилгидразинов. // Химия гетероцикл. соедин. - 1994. - № 5. - С.686-692.
7. Поддубный И.С., Беленький Л.И., Стручкова М.И., Краюшкин М.М. Спектры ЯМР 1Н и 13С 2,5-дизамещенных 1,3,4-оксадиазолов. // Химия гетероцикл. соедин. - 1994.- № 6.- С.834-842.
8. Поддубный И.С., Беленький Л.И., Краюшкин М.М. Природа восстановителя и механизм восстановительной конденсации трихлорметиларенов с гидроксиламином и гидразинами в пиридине. // Химия гетероцикл. соедин. - 1995. - № 6. - С.830-842.
9. Беленький Л.И., Поддубный И.С., Краюшкин М.М. Новая редокс-система: трихлорметиларен – пиридиновое основание. // Химия гетероцикл. соедин. - 1995. - № 10.- С.1373-1375.
10. Belen’kii L.I., Poddubny I.S., Krayushkin M.M. A New Redox System: Trichloro-methylarene - Pyridine Base. On the Mechanism of the Synthesis of N-(4-Pyridyl)pyridinium Dichloride. // Tetrahedron Letters. - 1995.- Vol. 36, № 28.- Р.5075-5078.
11. Поддубный И.С., Беленький Л.И., Краюшкин М.М. Взаимодействие трихлор-метиларенов с производными гидразина. Синтез 2,5-дизамещенных 1,3,4-оксадиазолов и 1,3,4-тиадиазолов. // Изв. АН, Серия химическая. - 1996.- № 5. - С.1246-1249.
12. Беленький Л.И., Луйксаар С.И., Поддубный И.С., Краюшкин М.М. Новые синтезы симметричных 2,5-диарил-1,3,4-оксадиазолов и 1,4-фениленбис-1,3,4-оксади-азолов. // Изв. АН, Серия химическая. - 1998. - № 11.- С.2309-2316.
13. Кузнецов А.А., Поддубный И.С., Ткачук Л.Н. Совместное получение ацетил-хлорида и бензальдегида из уксусной кислоты и бензальхлорида или кубовых остатков производства хлористого бензила. // Журнал прикладной химии. - 2000. - Т.73, вып.7.- С.1145-1148.
14. Поддубный И.С., Беленький Л.И., Краюшкин М.М. Реакции мезитотрихлорида с 2-амино- и 2-амино-5-бромпиридинами. // Химия гетероцикл. соедин. - 2000.- № 10.- С.1351-1353.
15. Беленький Л.И., Поддубный И.С., Луйксаар С.И., Краюшкин М.М. Некоторые реакции пиридиниевых солей, образующихся из трихлорметиларенов, с N- и C-нуклео-филами. // Химия гетероцикл. соедин.- 2000. - № 10. - С.1354-1359.
Статьи в реферируемом журнале и сборниках научных трудов
16. Поддубный И.С. Улучшенный метод радикального хлорирования алкиларо-матических и предельных углеводородов с получением продуктов, используемых в шинной промышленности и в производстве РТИ. // Мир шин. - 2008. - № 9, (52). - С.16-18.
17. Беленький Л.И., Поддубный И.С., Краюшкин М.М. Пути образования 1,2,4- и 1,3,4-оксадиазолов при восстановительной конденсации трихлорметиларенов с гидроксил-амином и гидразинами в пиридине. // Карбонильные соединения в синтезе гетероциклов. Сборник научн. трудов, Саратов: Изд-во СГУ, 1992. - ч.1. - С.36.
18. Беленький Л.И., Луйксаар С.И., Поддубный И.С., Краюшкин М.М. Новые синтезы 2,5-дизамещенных 1,3,4-оксадиазолов и 1,4-фениленбис-1,3,4-оксадиазолов. // Новые достижения в органической химии. Сборник научн. трудов, Саратов: Изд-во СГУ, 1997. - С.28.
Патенты на изобретения
19. Поддубный И.С., Кузнецов А.А., Мудрый Ф.В., Мильготин И.М. Способ получения гексахлорпараксилола. // Патент РФ № 2108317, заявлено 22.11.1996, опубл. 10.04.1998. Бюл. № 10.
20. Богач Е.В., Кузнецов А.А., Мильготин И.М., Мокрушин М.В., Поддубный И.С., Роик Ф.И., Сергеев А.А., Телегин И.В., Ткачук Л.Н., Мудрый Ф.В. Способ получения ацетилхлорида. // Патент РФ № 2135457, заявлено 24.04.1997, опубл.27.08.1999. Бюл. №24.
21. Поддубный И.С., Кузнецов А.А., Мудрый Ф.В., Вараксин В.В., Мильготин И.М., Усманов А.М., Богач Е.В., Головцов И.Н. Способ получения твердого хлорпарафина марки ХП-1100. // Патент РФ № 2136650, заявлено 21.04.1998, опубл. 10.09.1999. Бюл. №25.
22. Богач Е.В., Кузнецов А.А., Куликова О.А., Мильготин И.М., Мудрый Ф.В., Поддубный И.С., Гришин Б.С., Фроликова В.Г., Яловая Л.И., Патент РФ № 2142406, заявлено 21.04.1998, опубл. 10.12.1999. Бюл. № 34.
23. Поддубный И.С., Кузнецов А.А., Мильготин И.М., Мокрушин М.В., Петрухин В.Д. Способ получения бензальдегида. // Патент РФ № 2180329, заявлено 28.04.2000, опубл. 10.03.2002. Бюл. № 7.
24. Кузнецов А.А., Поддубный И.С., Богач Е.В., Мильготин И.М., Мудрый Ф.В., Гришин Б.С., Гончарова Л.Т., Пичугин А.М., Сафронова Л.В. Модификатор для резиновых смесей (варианты) и способ его получения (варианты) // Патент РФ № 2213108, заявлено 30.08.2000, опубл. 27.09.2003. Бюл. № 27.
25. Кутянин Л.И., Кузнецов А.А., Поддубный И.С., Иванова Н.А., Сергеев С.А. Способ стабилизации галогенированных парафинов. // Патент РФ № 2245318, заявлено 14.08.2002, опубл. 27.01.2005. Бюл. № 3.
Тезисы докладов
26. Беленький Л.И., Поддубный И.С., Краюшкин М.М. Конденсация трихлор-метиларенов с тиосемикарбазидом. // Сб. тезисов XVIII Конференции по химии и техноло-гии органических соединений серы. Казань, 1992. - С.25.
27. Belen’kii L.I., Poddubny I.S., Krayushkin M.M. Mechanism of Redox Condensation of Trichloromethylarenes with Hydroxylamine or Hydrazine in Pyridine. A Novel Synthesis of 1-(4-Pyridyl)pyridinium dichloride. // Abstracts of papers of XIV International Congress on Heterocycles Chemistry. - Antverpen, Belgium. - 1993, PO 3-169.
28. Belen’kii L.I., Poddubny I.S., Krayushkin M.M. Synthesis of 2-Amino-5-aryl-1,3,4-thiadiazoles from Trichloromethylarenes: The Effect of Reaction Conditions. // Abstracts of papers of 16th International Symposium on Chemistry of Organic Compounds of Sulfur, Merseburg. - 1994, О 3.10, Р.98.
29. Belen’kii L.I., Poddubny I.S., Krayushkin M.M. Synthesis of 1,3,4-Oxadiazoles and 1,3,4-Thiadiazoles from Trichloromethylarenes: The Effect of Reaction Conditions. // Abstracts of papers of 10th International Conference on Organic Synthesis. - Bangalore, India. - 1994. - P-FRI-61.
30. Belen’kii L.I., Poddubny I.S., Krayushkin M.M. A New Redox System: Trichloro-methylarene - Pyridine Base. // Abstracts of papers 14th International Congress on Heterocyclic Chemistry, August 6-19, 1995. Taipei International Convention Center. – 1995. - PO3-269.
31. Belen’kii L.I., Poddubny I.S., Krayushkin M.M. A New Redox System: Trichloro-methylarene - Pyridine Base. // Abstracts of papers Vth Symposium on Heterocyclic Chemistry Blue Danube, June 14-17, 1995. - Casta Paperniсka, Slovakia, 1995. - OP 12.
32. Беленький Л.И., Поддубный И.С., Краюшкин М.М. Новая редокс-система: трихлорметиларен – пиридиновое основание. // Сб. тезисов Межинститутского коллокви-ума “Химия азотистых гетероциклов”. Черноголовка, 1995. - С.6.
33. Belen’kii L.I., Luiksaar S.I., Poddubnyi I.S., Krayushkin M.M. New synthesis of 2,5-disubstituted 1,3,4-oxadiazoles and 1,4-phenylene-bis-1,3,4-oxadiazoles. // Electronic Conference on Heterocyclic Chemistry, Synthesis and Methodology, 29 June – 24 July 1998 (http//www.ch.ic.ac.uk/ectoc/echet98/ectoc mol.html).
34. Инжинова Л.М., Чеботарева Л.С., Худоян И.С., Кузнецов А.А., Поддубный И.С., Мудрый Ф.В. Исследование свойств модификаторов на основе гексахлорпара-ксилола и поливинилхлорида. // Сырье и материалы для резиновой промышленности. Настоящее и будущее. Тезисы докл. Пятой Российской научно-практической конференции резинщиков, Москва, 1998. - С.155-157.
35. Кузнецов А.А., Куликова О.А., Мудрый Ф.В., Поддубный И.С., Яловая Л.И., Фроликова В.Г. Разработка технологии получения полимерной серы на ОАО “Химпром” г. Волгоград. 1. Сравнительные испытания лабораторных образцов. // Сырье и материалы для резиновой промышленности. Настоящее и будущее. Тезисы докл. Пятой Российской научно-практической конференции резинщиков. Москва, 1998. - С.168, 169.
36. Беленький Л.И., Броховецкий Д.Б., Поддубный И.С., Луйксаар С.И., Краюшкин М.М. Гетероциклизация и восстановительная конденсация - конкурирующие реакции трихлорметиларенов с гидроксиламином и гидразинами. // Сб. тезисов 1-ой Всероссий-ской конференции по химии гетероциклов памяти А.Н.Коста. Суздаль, 2000. - С.19.
37. Поддубный И.С. Улучшенный метод радикального хлорирования алкиларома-тических и предельных углеводородов с получением продуктов, используемых в шинной промышленности и в производстве РТИ. // Резиновая промышленность. Сырье. Материалы. Технологии. Тезисы докл. XIV Международной научно-практической конференции. Москва. - 2008. - С.174-176.
38. Поддубный И.С. Новый эффективный способ получения высокостабильных галогенированных парафинов, используемых в производстве РТИ и полимерных композиций. // Сб. тезисов XIX Симпозиума “Проблемы шин и резинокордных композитов”. Москва, 2008. - том 2. - С.140-144.