

Радикальная химия карбонилов железа
РОССИЙСКАЯ АКАДЕМИЯ НАУК
ИНСТИТУТ ЭЛЕМЕНТООРГАНИЧЕСКИХ СОЕДИНЕНИЙ
им. А. Н. НЕСМЕЯНОВА
На правах рукописи
УДК 546. 725: 547.257.2:
547.489.2 + 541.124
Белоусов Юрий Анатольевич
Радикальная химия карбонилов железа
Специальность 02.00.08 — химия элементоорганических соединений
АВТОРЕФЕРАТ
диссертации на соискание учёной степени
доктора химических наук
Москва - 2008 г.
Работа выполнена в Институте элементоорганических соединений
им. А. Н. Несмеянова Российской Академии Наук (ИНЭОС РАН)
| Официальные оппоненты: | Черкасов Владимир Кузьмич, член-корреспондент РАН, д.х.н., ИМХ им. Г.А. Разуваева РАН, Н.-Новгород Устынюк Николай Александрович, д.х.н., проф., ИНЭОС РАН Мельников Михаил Яковлевич, д.х.н., проф., Химфак МГУ им. М.В. Ломоносова |
Ведущая организация: Институт общей и неорганической химии им. Н.С.
Курнакова РАН
Зашита состоится 2008 г. в часов на заседании Диссертационного Совета Д 002.250.01 по химическим наукам при Институте элементоорганических соединений имени А. Н. Несмеянова Российской академии наук по адресу: 119991, г. Москва, ГСП-1, В-334, ул. Вавилова, 28, ИНЭОС РАН
С диссертацией можно ознакомиться в библиотеке ИНЭОС РАН
Автореферат разослан 2008 г.
Учёный секретарь
Диссертационного Совета Д 002.250.01
кандидат химических наук Т. А. Ларина
ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ
Актуальность проблемы. Химия карбонилов переходных элементов возникла немногим более века назад. Решающее влияние на развитие этой области оказали процессы каталитической переработки синтез-газа (СО и водород), синтеза углеводородов по Фишеру-Тропшу и гидроформилирования, исследование которых привело к использованию карбонилов металлов в качестве гомогенных катализаторов (синтез Реппе) и замене синтез-газа на водяной газ (CO и H2O). Поскольку ключевым компонентом газовых смесей является CO, природа действия таких катализаторов должна определяться закономерностями химии карбонилов металлов. Интенсивность исследований в области С1–химии, основанной на сырье, альтернативном нефти, напрямую зависит от природных запасов углеводородов. По мере истощения запасов нефти с необходимостью встанет вопрос о переходе нефтехимического комплекса на процессы переработки CO в углеводороды и другие органические продукты. Однако, несмотря на отдельные успешно реализованные процессы конверсии CO, в большинстве случаев, по экономическим причинам, они пока не могут составить конкуренцию процессам нефтехимии. Поэтому поиск недорогих катализаторов, способных в мягких условиях работать селективно, является актуальной задачей. Для ее решения необходимы глубокие исследования поведения различных координационных соединений в реакциях с CO. Наиболее перспективными из них являются комплексы карбонилов железа (КЖ), интерес к которым определяется их доступностью, стабильностью и высокой активностью в различных реакциях. Таким образом, проведение исследований в области химии КЖ является актуальной задачей и представляет значительный интерес как для фундаментальной координационной и металлоорганической химии, так и для решения прикладных задач, связанных с созданием гомогенных катализаторов и новых материалов, обладающих ценными свойствами.
Целью данной работы является исследование реакционной способности карбонилов железа в условиях протекания радикальных и ион-радикальных реакций.
Научная новизна. Разработан общий подход к описанию взаимодействий карбонилов металлов с основаниями Льюиса в рамках схемы редокс-диспропор-ционирования, включающей стадию активирующего комплексообразования с последующим одноэлектронным переносом с активированного донором электронов карбонильного комплекса на другую молекулу карбонила металла. Предложена схема каталитического процесса восстановительного карбонилирования нитро- и нитрозосоединений в присутствии карбонилов железа, в котором роль каталитически активных частиц выполняют железокарбонильные анион-радикалы Fe2(CO)8, Fe3(CO)11 и Fe4(CO)13. Показано, что этот каталитический цикл включает превращения через 17 19 17 комплексы, и весь процесс протекает в радикально-цепном режиме. Выявленные закономерности позволяют использовать их для поиска на основе карбонилов металлов эффективных катализаторов процессов карбонилирования при контролируемом электрохимическом потенциале среды.
Практическая ценность. В результате проведенных исследований разработан эффективный способ получения комплексов бис(алкилтиотрикарбонилжелеза), используемых в качестве катализаторов некоторых процессов карбонилирования, а также метод получения нитевидных монокристаллов -железа при термической диссоциации карбонилов железа в газовой фазе. Предложена методика измерения концентрации железокарбонильных анион-радикалов во времени с использованием метода остановленной струи.
Апробация работы. Результаты диссертационной работы докладывались на I (Москва, 1979 г.), II (Горький, 1982 г.) и IV (Казань, 1988 г.) Всесоюзных конференциях по металлоорганической химии, VI Всероссийской конференции по металлоорганической химии (Нижний Новгород, 1995 г.), IV Всесоюзной конференции по химии азотсодержащих гетероциклических соединений (Новосибирск, 1987 г.), на II Разуваевских чтениях (Нижний Новгород, 1995 г.), на IV (Чехословакия, 1982 г.), VIII (Венгрия, 1989 г.) и X (Греция, 1993 г.) Европейских (FECHEM) конференциях по металлоорганической химии, IV Международном симпозиуме по гомогенному катализу (Ленинград, 1984 г.), VI Международной конференции по органическому синтезу(IUPAC), (Москва, 1986 г.), XXVIII Международной конференции по координационной химии (ГДР, 1990 г.), Международном симпозиуме «Достижения науки в органической химии» (Украина, 2006 г.), Международной конференции посвящённой 50-тилетию ИНЭОС РАН им. А. Н. Несмеянова (Москва, 2004 г.)
Публикации. По теме диссертации опубликовано 37 работ в отечественных и международных изданиях, в том числе 1 обзор и 2 авторских свидетельства.
Объём и структура работы. Диссертация изложена на 231 стр. и включает 19 таблиц, 25 схем и 42 рисунка; состоит из введения, обзора, обсуждения полученных результатов (4 главы), экспериментальной части, выводов и списка литературы (439 наименований).
Во введении рассмотрена перспектива развития гомогенного катализа на основе карбонильных комплексов металлов в области С1–химии, основанной на сырье, альтернативном нефти.
В литературном обзоре изложено современное состояние исследований области в химии карбонилов железа и основные схемы их превращений.
В экспериментальной части рассмотрены способы получения N-ферроценилазолов, тиокарбонильных комплексов железа, а также нитевидных кристаллов -железа. Представлены методики получения различных радикальных железокарбонильных комплексов. Разработана методика измерения концентрации железокарбонильных анион-радикалов во времени с использованием метода остановленной струи. Приведена методика расчёта констант скоростей реакций, и получено решение обратной кинетической задачи для схемы, включающей одноэлектронное редокс-инициирование с предварительным активирующим комплексообразованием.
Исследование выполнено в Институте элементоорганических соединений
им. А. Н. Несмеянова Российской Академии Наук в лаборатории синтеза металлоорганических соединений и лаборатории механизмов химических реакций как плановая тема института по направлению «Синтез, исследование строения, реакционной способности и практически важных свойств металлоорганических и координационных соединений» при финансовой поддержке Российского Фонда Фундаментальных исследований (гранты № 95-03-09451 и № 98-03-32227) и обобщает цикл исследований за период 1979 - 2007 гг.
Основное содержание работы
1. Взаимодействие карбонилов железа с основаниями Льюиса
При исследовании реакции карбонилов железа (КЖ) с натриевыми солями азолов (AzNa) непосредственно в резонаторе ЭПР-спектрометра мы впервые обнаружили образование железокарбонильных анион-радикалов (ЖКАР) Fe2(CO)8 (2), Fe3(CO)11(3), Fe3(CO)12 (4) и Fe4(CO)13 (5). Дальнейшие исследования взаимодействия КЖ Fe(CO)5, Fe2(CO)9 и Fe3(CO)12 с различными основаниями Льюиса, взятыми как в анионной, так и нейтральной формах, показали, что ЖКАР Fe(CO)4 (1) и 2-5 легко генерируются уже на ранних стадиях при малых глубинах превращений КЖ. При этом концентрации некоторых из них достигают сравнительно больших величин (10-5 – 10-2 моль·л -1). Эти данные можно было интерпретировать как результат протекания процессов одноэлектронного восстановления исходных КЖ, по аналогии с электрохимическим восстановлением или действием сильных восстановителей типа натриевого зеркала и бензофенонкетила натрия. Однако рассматривать эти основания Льюиса в качестве восстановителей по термодинамическим соображениям вряд ли уместно, так как потенциал окисления предполагаемого донора слишком высок. Например, в случае L = Cl, OH он составляет в ацетонитриле +2.24 В и +0.92 В соответственно.

Эти процессы следует отнести к «неправильным реакциям, идущим против потенциала», аналогичным исследованному Педерсеном (1973) взаимодействию пара-хинонов с основаниями. Для объяснения механизмов реакций такого типа Абакумовым (1975) была предложена концепция «активирующего комплексообразования», которая, применительно к КЖ, заключается в следующем. Если донор (основание Льюиса, например, в анионной форме L) непосредственно не может восстановить акцептор КЖ (С), то образуется комплекс CL, который приобретает свойство восстановителя по отношению к молекуле КЖ, и одноэлектронный перенос уже осуществим:

Схема 1
Для реакций КЖ с основаниями Льюиса первая стадия процесса – активирующее комплексообразование – заключается в присоединении основания по карбонильной группе с образованием известных из литературы аддуктов, где L = H, OR, NR2 (R = H, Alk),

ИК-спектроскопическое изучение реакций пентакарбонила железа (ПКЖ) с тиолятами натрия в ТГФ показало образование таких аддуктов и с S-основаниями Льюиса. В этих системах на первых стадиях процесса при избытке тиолята появляется полоса поглощения карбонильной группы «ацильного» типа в области 1620–1670 см-1. В таблице 1 приведены данные ИК-спектров в области поглощения карбонильных групп для комплексов Na+[Fe(CO)4C(O)L] в сравнении с известным метоксильным комплексом.
Таблица 1. Данные ИК-спектров комплексов Na+[Fe(CO)4C(O)L]
| L | (CO), см-1, (ТГФ) | ||||
| C2H5S | 2031 | 1959 | 1913 | 1900 | 1661 |
| n-C4H9S | 2022 | 1941 | 1912 | 1899 | 1651 |
| t-C5H11S | 2023 | 1931 | 1904 | 1889 | 1621 |
| CH3O | 2023 | 1929 | 1913 | 1901 | 1585 |
Для доказательства второй окислительно-восстановительной стадии схемы 1 в аналитически чистом виде была получена соль (PPN)[Fe(CO)4C(O)OMe] (PPN+ - катион бис-(трифенилфосфин)иминия), которая так же, как и исходный ПКЖ, не даёт сигналов в спектре ЭПР. Проведение реакции метоксикарбонилтетракарбонилферрата с ПКЖ непосредственно в резонаторе ЭПР-спектрометра показало, что происходит быстрое восстановление КЖ с образованием ЖКАР даже при температурах ниже 80 °C. При этом оказалось, что вид ЭПР-спектра и характер его изменения во времени такой же, как и в исходной реакции Fe(CO)5 с метилатом натрия (рис. 1).
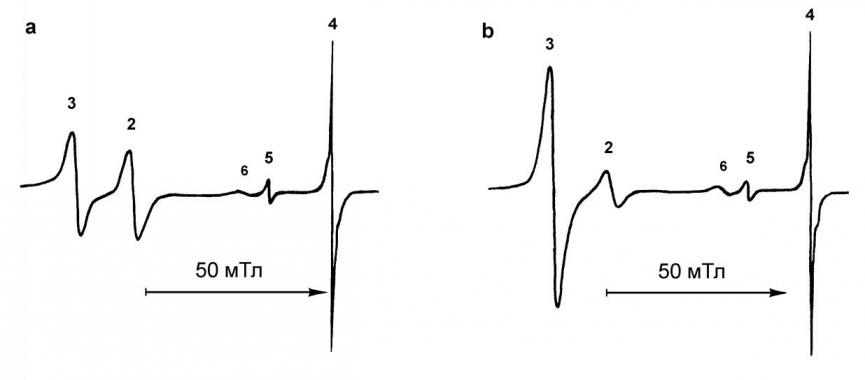
Рис. 1. Спектры ЭПР, наблюдаемые через 5 минут после начала реакции: a. Fe(CO)5 с PPN[Fe(CO)4C(O)OCH3] в ТГФ при -80 °C; b. Fe(CO)5 с CH3ONa в ТГФ при -80 °C. Сигналы от ЖКАР 2 – Fe2(CO)8 (g = 2.0386), 3 – Fe3(CO)11 (g = 2.0497), 4 – Fe3(CO)12 (g = 2.0013), 5 – Fe4(CO)13 (g = 2.0134); 6 – сигнал от радикала с g = 2.0182.
Таким образом, взаимодействие КЖ с анионными основаниями Льюиса может быть представлено в следующем виде:

Эта схема инициирования имеет достаточно универсальный характер и реализуется в реакциях c Fe(CO)5, Fe2(CO)9 и Fe3(CO)12 с широким кругом анионных оснований Льюиса в виде натриевых солей NaL, (L = OR, SR, NR2, CR2NO2 (R = H, Alk, Ph)), солей щелочных металлов (L = H, F, Cl, Br, I, CN, SCN, N3, MeCO2, CF3CO2, MeSO3, NO2) и комплексных катионов Et4N+ (L = Cl, Br), n-Bu4N+ (L = I), PPN+ (L = Cl, CN, NO2), а также натриевых солей двухзарядных анионов S2, CO32, SO42. В качестве растворителей использовали ТГФ, метилтетрагидрофуран, диэтиловый эфир, моноглим, 1,4-диоксан, хлористый метилен, метанол, этанол и смесь ТГФ – вода (10:1 об.). ЖКАР 2 – 5 образуются также при взаимодействии Fe5C(CO)15 с натриевыми производными анионов OH, OMe, OEt, SEt, NEt2, Cl и H.
Редокс-диспропорционирование КЖ может происходить, но значительно медленнее, даже в случае нейтральных оснований Льюиса. В частности, реакции Fe(CO)5 с основаниями (ROH, RSH, R2NH) хотя и приводят к образованию набора радикалов, практически идентичного тому, который получается в реакциях с участием анионных форм, однако протекают намного медленнее и лишь при повышенных температурах. Меньшая скорость генерирования ЖКАР может быть связана как с низкими константами равновесия первой стадии активирующего комплексообразования (уравнение 4), так и с затратой энергии на генерирование и разделение зарядов на второй редокс-стадии (уравнение 5):
 Кроме перечисленных выше, были исследованы реакции КЖ с традиционными растворителями и реагентами, обладающими донорными свойствами и представляющими собой нейтральные основания Льюиса, такими как, пиридин, ДМФА, ДМСО, а также o-фенантролин и триметиламиноксид.
Кроме перечисленных выше, были исследованы реакции КЖ с традиционными растворителями и реагентами, обладающими донорными свойствами и представляющими собой нейтральные основания Льюиса, такими как, пиридин, ДМФА, ДМСО, а также o-фенантролин и триметиламиноксид.
Исследование скорости реакций оснований Льюиса с Fe(CO)5, Fe2(CO)9 и Fe3(CO)12 показало, что легче всего реагирует Fe3(CO)12, а труднее - Fe(CO)5. Это обусловлено тем, что потенциал восстановления Fe3(CO)12 E red = - 0.32 - 0.44 В намного выше, чем у Fe(CO)5 E red = - 1.77 В. Поэтому стадия электронного переноса (уравнения 3 и 5) легче реализуется в первом случае. По-видимому, гетеро-генный характер систем Fe2(CO)9/L (или L) в данном случае не вносит решающего вклада в скорость процесса. При этом перенос электрона с активированного комплекса на соседнюю молекулу КЖ на второй стадии (уравнения 3 и 5) происходит на поверхности кристалла и сопровождается делокализацией заряда по кристаллу, что уменьшает энергетические затраты и компенсирует диффузионные ограничения.
Таким образом, схему, включающую стадию активирующего комплексообразования с последующим одноэлектронным переносом электрона с активированного комплекса на другую молекулу карбонила металла, можно рассматривать как достаточно универсальную для реакций диспропорционирования КЖ с основаниями Льюиса.
Образующиеся по уравнениям (3), (5) комплексы железа формально имеют степени окисления (+I) и (-I) и инициируют развитие двух цепей превращений, обусловленных реакциями замещения в координационной сфере радикалов. ЖКАР, являясь достаточно стабильными радикалами, легко детектируются, тогда как регистрация нестабильных радикалов Fe(+I) (конфигурация атома железа d7) представляет собой сложную задачу. В том случае, если L - жёсткое основание, происходит дестабилизация координационной сферы карбонильного комплекса с отщеплением CO-лигандов и дальнейшим окислением железа до Fe2+. В итоге образуются неорганические соединения типа FeL2 (если основание L – анион), например Fe(Az)2, или FeCl2 или соли катиона [FeLk]2+ (если основание находится в нейтральной форме). В тех случаях, когда L – мягкое основание, возможна стабилизация координационной сферы в рамках железокарбонильного комплекса Fe(+I). Помимо этого возможны случаи, когда основание превращается из жёсткого в более мягкое, например из NO2 в NO, либо происходит увеличение -связанности лиганда L с металлоостовом. Такие радикалы Fe(+I) в отдельных случаях удаётся зафиксировать методом ЭПР или непосредственно, или в форме спиновых аддуктов.
При проведении реакций Fe(CO)5 с натриевой солью аци-формы нитрометана (нитроната) непосредственно в резонаторе ЭПР-спектрометра при – 80°C наблюдается образование ЖКАР 2 – 5, а также радикалов с g = 2.0182 (6) и g = 2.0461 (уширенные синглеты) и нитрозильных радикальных комплексов Fe(CO)3NO(7) (g = 2.0327, aN = 2.1 мТл), Fe(NO)2(THF)2+ (8) (g = 2.0217, aN = 1.6 мТл), Fe(NO)3CO (9) (g = 1.9877, aN = 5.0 мТл). Идентификацию нитрозильных радикалов осуществляли на основании реакций встречного синтеза при окислении гексафторфосфатом феррициния (FcPF6) соответствующих диамагнитных аналогов или литературных данных. Радикал Fe(CO)3NO был зафиксирован также в виде спинового аддукта (CO)3(NO)FeN(O)-t-Bu (g = 2.0040, aN = 16.8 мТл, aN = 0.65 мТл).
Реакции Fe(CO)5 с солями нитрита оказались по набору радикальных продуктов близки к реакциям с нитронатами. Образование и расходование ЖКАР 2 - 5, а также радикалов 6 – 9 одинаково для обеих реакций. Помимо них были идентифицированы радикальные комплексы Fe(CO)2(NO)2+ (10) (g = 2.0286, aN = 2.8 мТл) и Fe3(NO)8+ (11). Строение первого было доказано встречным синтезом в реакции Fe(CO)2(NO)2 с FcPF6, а второго – по литературным данным.
Таким образом, пути превращений комплексов Fe(+I), образующихся в реакциях КЖ с нитритами и нитронатами в результате одноэлектронного редокс-диспропорционирования (уравнение (3)), определяются: 1) формированием координационной сферы атома железа путём замены CO на NO лиганды, которые являются более мягкими, чем исходные нитрит- и нитронат-анионы; 2) образованием кластерных структур, стабилизированных делокализацией неспаренного электрона по всему металлоостову.
Благоприятная ситуация для обнаружения радикальных комплексов Fe(+I) складывается в реакциях КЖ с гидросульфидом и тиолятами натрия, которые можно отнести к мягким основаниям Льюиса. В этих случаях наблюдается образование тех же ЖКАР 2 – 5, а также радикальной частицы 6. В самом начале реакций Fe(CO)5 с тиолятами и гидросульфидом натрия в спектрах ЭПР удаётся детектировать широкие сигналы короткоживущих радикалов (12) с g = 2.0463 (L = S-n-C4H9), g = 2.0459 (L = SH), g = 2.0465 (L = S-t-C5H11), которые можно рассматривать как одни из первичных парамагнитных продуктов окисления анионного комплекса [Fe(CO)4C(O)SR] в уравнении (3), например [Fe(CO)4C(O)SR] или [Fe(CO)4SR]. Затем в спектрах ЭПР возникают сигналы анион-радикалов Fe2(CO)6(SR)2 (13) с g-фактами: 2.0337 (R = Et), 2.0336 (R = n-Bu), 2.0333 (R = n-C8H17), 2.0338 (R = SH), 2.0346 (R = Ph), и анион-радикалов Fe3(CO)9(SR)2 (14) (g = 2.0114 при R = n-Bu, t-C5H11; g = 2.0115 при R = Et; g = 2.0116 при R = n-C8H17). Эти радикалы идентифицированы на основании реакций встречного синтеза при восстановлении диамагнитных аналогов Fe2(CO)6(SR)2 и Fe3(CO)9(SR)2 на натриевом зеркале.
Образование в реакции Fe(CO)5 с тиолятами и гидросульфидом натрия радикалов карбонильных комплексов Fe(+I) можно представить схемой превращений, в которой изменение ядерности радикальных комплексов происходит в результате процессов обмена лигандов CO на исходный КЖ или анион L = SR в координационной сфере первоначально образующихся по реакции (3) (n = 1, m = 5) комплексов Fe(+I) (уравнения 6 - 7)

Последующее окисление анион-радикалов 13 и 14, содержащих два тиолятных лиганда, в результате катализа и замещения посредством электронного переноса (ETC) или при дальнейшем диспропорционировании (уравнение 8) приводит к образованию конечных диамагнитных продуктов, например соединений Fe2(CO)6(SR)2, Fe3(CO)9(SR)2 и солей карбонилферрат-анионов (КФА).

Соответственно, при восстановлении радикалов, содержащих один тиолятный лиганд (верхняя строка уравнения 7), могут получаться диамагнитные анионы Fe(0) типа [Fe(CO)4SR], [Fe2(CO)7SR] и [Fe3(CO)9SR].
В реакциях КЖ с основаниями Льюиса на стадии одноэлектронного редокс-диспропорционирования (3), (5) образуются неустойчивые (за исключением Fe3(CO)12) электроноизбыточные ЖКАР Fen(CO)m (n = 1, 2; m = 5, 9), которые переходят при декарбонилировании в более устойчивые электрон-дефицитные ЖКАР Fen(CO)m-1. В терминах правила Сиджвика о замкнутой 18-электронной конфигурации, осуществляется переход от 19 к 17 комплексам:

Как правило, образование ЖКАР Fe2(CO)8, Fe3(CO)11, Fe3(CO)12 и Fe4(CO)13 наблюдается при любых сочетаниях исходного КЖ и основания. Различия проявляются лишь в очерёдности их возникновения и расходования на разных стадиях процесса. Например, в реакциях Fe(CO)5 с основаниями Льюиса ЖКАР появляются в такой последовательности: Fe(CO)4 (1), Fe2(CO)8(2), Fe3(CO)12 (4) и Fe3(CO)11(3), Fe4(CO)13(5). Моноядерный анион-радикал Fe(CO)4 (1) удалось обнаружить на первых стадиях реакции Fe(CO)5 с сильными основа-ниями, такими как NaOMe и NaOEt, при -100°C в метилтетрагидрофуране, т.е. сначала образуется моноядерный ЖКАР, а затем последовательно би-, три- и тетраядерные анион-радикалы. В реакциях c Fe3(CO)12 сначала возникают трёхъ-ядерные анион-радикалы Fe3(CO)11, Fe3(CO)12, а затем уже биядерный Fe2(CO)8 и тетраядерный Fe4(CO)13. В гетерогенной системе с Fe2(CO)9 сначала генерируется биядерная частица Fe2(CO)8, а на более поздних стадиях появляются Fe3(CO)11, Fe3(CO)12 и Fe4(CO)13. Аналогичная последовательность генерирования ЖКАР наблюдалась и при восстановлении КЖ на натриевом зеркале. Если процесс идёт в замкнутой системе, например в запаянных ампулах, то на более поздних стадиях при комнатной температуре между ЖКАР Fe2(CO)8, Fe3(CO)11, Fe3(CO)12 и Fe4(CO)13 устанавливается равновесие с преобладанием трёхъядерного анион- радикала Fe3(CO)11. Последовательные взаимопревращения ЖКАР с учётом того, что координационная сфера анион-радикалов Fe(CO)4, Fe2(CO)8, Fe3(CO)12, Fe3(CO)11 и Fe4(CO)13 достаточно лабильна и способна к быстрому лигандному и электронному обмену, могут быть описаны обратимыми реакциями замещения CO-групп на Fe(CO)5:

Для ответа на основной вопрос, лежат ли радикальные продукты, полученные при взаимодействии карбонилов железа с основаниями Льюиса на координате реакции или являются побочными, была исследована кинетика накопления и расходования промежуточных железокарбонильных анион-радикалов в модельной реакции додекакарбонилтрижелеза с этилтиолятом в ТГФ при 19,5°C, для которой известны как промежуточные радикальные частицы, так и конечные продукты. Реакцию проводили непосредственно в резонаторе ЭПР-спектрометра в проточной системе с использованием метода остановленной струи. На рисунке 2 изображены кривые временной зависимости амплитуды сигналов анион-радикалов Fe3(CO)12 и Fe3(CO)11. Аналогичные зависимости скоростей накопления и расходования железокарбонильных анион-радикалов Fe3(CO)12 и Fe3(CO)11 наблюдались и в реакциях Fe3(CO)12 с NaOEt и NaOH.
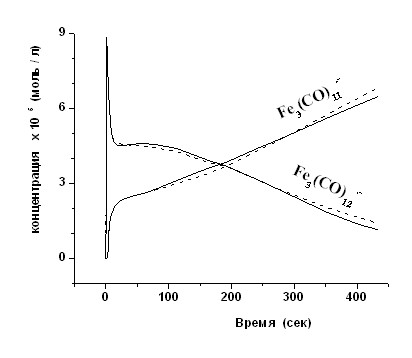 | Рис. 2. Временная зависимость концентрации сигналов ЭПР Fe3(CO)12 и Fe3(CO)11 в реакции Fe3(CO)12 ([C]0 = 3.79·10-3 моль·л-1) с (Et4N)SEt ([C]0 =1.09·10-3 моль·л-1) в ТГФ. Непрерывные линии – экспериментальные данные, пунктирные – расчётные данные. |
На первых стадиях процесса идёт интенсивное генерирование Fe3(CO)12 с последующим быстрым расходованием. При этом концентрация Fe3(CO)11 успевает вырасти незначительно. Такое поведение хорошо укладывается в рамки предложенного нами механизма двухстадийного редокс-инициирования (уравнения 2 и 3).

Схема 2
Полученные зависимости концентрации от времени для обоих анион-радикалов вместе с концентрацией исходных реагентов закладывались в расчётную
программу, в основу которой была заложена схема 2, включающая стадию активи-рующего комплексообразования с последующим одноэлектронным переносом и дальнейшими реакциями в двух относительно независимых цепях превращений, обусловленных реакциями замещения в координационной сфере ЖКАР и комплексов Fe(+1). Решение обратной кинетической задачи для заданной схемы заключалось в определении констант скоростей реакций, при которых расчётные кинетические кривые накопления и расходования (пунктирные линии на рис. 2) для обоих анион-радикалов совпадают с полученными в эксперименте (непрерывные линии).
В таблице 2 представлен один из возможных наборов констант скоростей для реакций (1) - (16), при которых достигается удовлетворительное совпадение экспериментальных и рассчитанных по схеме 2 концентраций анион-радикалов Fe3(CO)12 и Fe3(CO)11 (см. рис. 2). Важным моментом является то, что схема 2 носит достаточно полный характер и применима для описания реакций карбонилов железа с различными анионными основаниями Льюиса. Например, если исходным является Fe(CO)5, то процесс начинается со стадий (12)-(15), при этом уравнения (1), (2), (5) и (6) не учитываются.
Таблица 2. Значения констант скоростей процесса взаимодействия Fe3(CO)12 с (Et4N)SEt в ТГФ. (Размерность для мономолекулярных реакций с-1, для бимолекулярных моль·л-1·с-1).
| k1 = 8.34·103 | k-1 = 8.68·101 | k2 = 1.15·102 | k-2 = 5.00·10-1 |
| k3 =5.85·10-1 | k-3 =1.55·103 | k4 = 5.78·103 | k-4 = 1.60·104 |
| k5 = 2.68·101 | k6 = 2.98·101 | k7 = 5.36·104 | k8 = 9.75·107 |
| k9 = 1.46·101 | k10 = 1.68·101 | k-10 = 5.55·102 | k11 = 9.07·102 |
| k-11 = 8.81·105 | k12 = 2.95·104 | k-12 = 1.14·101 | k13 = 9.05·100 |
| k14 = 9.57·104 | k15 = 1.04·105 | k16 = 6.05·10-3 |
Таким образом, взаимодействие карбонилов железа с основаниями Льюиса является радикально-цепным процессом, на первых стадиях которого происходит одноэлектронное редокс-инициирование с предварительным комплексообразованием. Дальнейшие превращения обусловлены электронным и лигандным обменом в координационной сфере семнадцатиэлектронных координационно-ненасыщенных частиц.
2. Взаимодействие карбонилферрат- и гидридокарбонилферрат-анионов с кислотами Бренстеда и Льюиса
Изучено взаимодействие карбонилферрат-анионов (КФА) и их протонированных аналогов - гидридокарбонилферрат-анионов (ГКФА): Na2Fe(CO)4·1.5(diox) (diox - 1,4-диоксан), Na2[Fe2(CO)8], (PPN)2[Fe2(CO)8], (PPN)2[Fe3(CO)11], (PPN)2[Fe4(CO)13], KHFe(CO)4, (PPN)[HFe(CO)4], (PPN)[HFe3(CO)11], (Et3NH)[HFe3(CO)11] с уксусной, трифторуксусной и серной кислотами. При взаимодействии этих анионов с кислотами Бренстеда в спектрах ЭПР наблюдаются интенсивные сигналы ЖКАР: Fe2(CO)8 (2), Fe3(CO)11 (3), Fe4(CO)13 (5) и Fe3(CO)12 (4). При этом, согласно данным ГХ-анализа, происходит выделение водорода. Последовательность возникновения ЖКАР в реакциях моно- и биядерных КФА и ГКФА следующая: сначала образуется анион-радикал 2, а затем 3, 5 и 4. В реакциях солей трехъядерных Fe3(CO)112- и HFe3(CO)11– первым образуется анион-радикал 3, а затем 4, 2 и 5. Важно отметить, что в спектрах ЭПР отсутствуют сигналы, соответствующие радикалам HFen(CO)m-1· (n = 1-3; m = 5, 9, 12).
В реакциях КФА и ГКФА с галогеналкилами RX (R = Me, Et, n-Bu; X = Cl, Br, I), PhCH2Cl, а также галогенангидридами кислот (R = Ac, Bz; X = Cl, Br) наибольшую активность проявляют соли Na2Fe(CO)4·1.5(diox), Na2[Fe2(CO)8], (PPN)2[Fe2(CO)2], KHFe(CO)4 и (PPN)[HFe(CO)4]. В спектрах ЭПР при –60°C в ТГФ наблюдается интенсивный рост сигналов ЖКАР Fe2(CO)8, Fe3(CO)11, Fe3(CO)12, Fe4(CO)13. При использовании (PPN)2[Fe3(CO)11], (PPN)2[Fe4(CO)13], (PPN)[HFe3(CO)11] и (Et3NH)[HFe3(CO)11] малоинтенсивные сигналы ЖКАР присутствуют лишь в спектрах ЭПР систем с более активными галогенангидридами карбоновых кислот.
При взаимодействии наиболее активных моноядерных анионов Fe(CO)42- и HFe(CO)4– с хлорангидридами RCl (R = Ac, Bz), помимо ЖКАР, образуются также ацилсодержащие железокарбонильные радикалы. Например, в спектре ЭПР реакционной смеси HFe(CO)4– с RCl при –60°C на первых стадиях процесса регистрируются дублеты с g = 2.0370, aH = 26.0 мТл для R = Bz и g = 2.0377, aH = 22.1 мТл для R = Ac, которым можно приписать строение R(H)Fe(CO)3О (14). Эти радикалы отличаются от известных HFen(CO)m-1·. При взаимодействии с бензоилхлоридом наблюдается также синглет с g = 2.0413 (рис. 3).
 | Рис. 3. ЭПР-спектр, наблюдаемый при взаимодействии PPN[HFe(CO)4] с BzCl в ТГФ при 60 °C через 4 мин после начала реакции. ЖКАР: a) Fe2(CO)8О, b) Fe3(CO)12О. Бензоилсодержащие железокарбонильные радикалы: c) Bz(H)Fe(CO)3О (14) ( g = 2.0370, aн= 26.0 мТл) и d) g = 2.0413. |
На ранних стадиях реакции Na2Fe(CO)4·1.5(diox) с бензоилхлоридом в метилтетрагидрофуране при –90°C в спектре ЭПР происходит рост интенсивности сигнала с g = 2.0489 (H = 6.1 мТл), которая, пройдя через максимум, падает, и уже на более поздних стадиях в этой области появляется сигнал, отвечающий анион-радикалу Fe3(CO)11О. По литературным данным, первоначальный сигнал соответствует радикалу PhCOFe(CO)4• (15).
В аналогичной реакции с ацетилхлоридом на первых стадиях процесса, наряду с Fe2(CO)8О (2) и Fe3(CO)12О (3), образуются ацетилсодержащие радикалы с g = 2.0436, g = 2.0342 и g = 2.0273 (рис. 4).
 | Рис. 4. ЭПР-спектр, наблюдаемый при взаимодействии Na2Fe(CO)4·1.5(diox) c AcCl в ТГФ при 60° C через 8 мин после начала реакции. ЖКАР: a) Fe2(CO)8, b) Fe3(CO)11, c) Fe3(CO)12, d) Fe4(CO)13, e) g = 2.0180. Ацетилсодержащие железокарбонильные радикалы: f) AcFe(CO)4• (15) (g = 2.0436), g) g = 2.0342 и h) g = 2.0273. |
Доказательством ацетильной природы этих радикалов может служить их получение по модельной реакции окисления комплекса (PPN)[MeCOFe(CO)4] гексафторфосфатом феррициния в ТГФ при –80°C. Образование (RCO)2Fe2(CO)6 связано с одноэлектронным окислением RCOFe(CO)4– до соответствующих радикалов с последующей димеризацией. Поэтому можно предположить, что эти ацетилсодержащие радикалы являются промежуточными частицами в процессе формирования биядерных анион-радикальных комплексов (уравнения (32) и (33)), которые при дальнейшем одноэлектронном окислении дают известные диамагнитные продукты.

Таким образом, реакции КФА и ГКФА с кислотами Бренстеда и Льюиса представляют собой редокс-процессы, в которых происходит одноэлектронное окисление КФА и ГКФА с образованием ЖКАР. Однако по термодинамическим причинам эти кислоты не могут быть окислителями. Для объяснения редокс-превращений в системе КФА (или ГКФА) + кислота Бренстеда (Льюиса), также как и для реакций КЖ с основаниями Льюиса, можно предложить схему «активирующего комплексообразования», с той лишь разницей, что в качестве донора B выступает КФА и ГКФА, а в качестве акцептора — их комплекс с кислотой BA.

Схема 3
Если донор B (КФА или ГКФА) непосредственно не может восстановить акцептор - кислоту (A+), но возможно образование их комплекса BA, то с участием такого комплекса одноэлектронный перенос уже осуществим. В рамках схемы 3 стадией активирующего комплексообразования являются известные процессы: либо протонирование КФА и ГКФА, либо окислительное присоединение к ним алкил- и ацилгалогенидов.

На примере моноядерного ГКФА (уравнение 16) показано, что стадией активирующего комплексообразования является протонирование. Для доказательства того, что электронный перенос происходит на следующей стадии (уравнение 17), необходимо было иметь чистые исходные комплексы, не содержащие следов продуктов окисления. Это удалось осуществить, путём дистилляции в вакууме H2Fe(CO)4 непосредственно в ампулу, помещённую в резонатор спектрометра ЭПР. В спектре ЭПР раствора H2Fe(CO)4 в метилтетрагидрофуране не обнаружено никаких сигналов при температуре ниже –60°С. То же наблюдается для соли [HFe(CO)4], растворённой в метилтетрагидрофуране, в широком интервале температур (вплоть до комнатной). В то же время, взаимодействие H2Fe(CO)4 с (PPN)[HFe(CO)4] в растворе метилтетрагидрофурана приводит к появлению сигналов ЖКАР уже при –100°С. На начальных стадиях процесса в спектрах ЭПР регистрируется интенсивный рост сигнала Fe2(CO)8. Затем появляются сигналы других анион-радикалов Fe3(CO)11, Fe4(CO)13 и Fe3(CO)12.

Реакции HFe(CO)4 с галогенорганическими соединениями могут быть представлены следующим образом: стадия активирующего комплексообразования — реакция (21), одноэлектронный перенос — реакция (22)

Образовавшийся неустойчивый 19-электронный комплекс RHFe(CO)4 переходит в более устойчивое 17 состояние за счёт отщепления RH или CO (реакция 23). В последнем случае возникают анион-радикальные частицы RHFe(CO)3 (14), которые обнаружены в реакции ацилгалогенидов RX (R = Ac, Bz) с HFe(CO)4.

Появляющийся на стадии (23) очень активный анион-радикал Fe(CO)4 – координационно ненасыщен, вследствие чего он имеет лабильную координационную сферу, в которой может происходить быстрый лигандный и электронный обмен. Важное следствие этого - присоединение кислоты Бренстеда или Льюиса к ЖКАР. Эта реакция является ключевым звеном многих процессов восстановления и карбонилирования.

Затем, по аналогии с реакцией (18), происходит образование анион-радикала Fe2(CO)8.

Дальнейшие превращения ЖКАР с повышением их ядерности происходят в радикально-цепном режиме:

На заключительных стадиях взаимодействия КФА и ГКФА с кислотами Бренстеда и Льюиса происходит восстановление ЖКАР 3 и 5 до соответствующих КФА в обменных реакциях типа (28), так как редокс-потенциалы этих анионов таковы, что эти процессы термодинамически выгодны.

Таким образом, конечные продукты реакции - это диамагнитные соединения, в основном представляющие собой модифицированные органические лиганды и полиядерные КФА и ГКФА.
3. Взаимодействие карбонилов железа с карбонилферрат-анионами
Взаимодействие [Fe(CO)4]2- и [Fe2(CO)8]2- с Fe(CO)5 и Fe3(CO)12 в ТГФ даже при пониженной температуре (80°C) идёт очень легко, причём уже на первых стадиях образуется обычный набор ЖКАР Fe2(CO)8, Fe3(CO)11, Fe3(CO)12 и Fe4(CO)13. Реакции [Fe(CO)4]2- и [Fe2(CO)8]2- с Fe2(CO)9, из-за плохой растворимости в ТГФ обоих исходных компонентов, протекают только при комнатной температуре с образованием обычного набора ЖКАР. В реакциях с участием солей трёх- и четырёхъядерных КФА [Fe3(CO)11]2- и [Fe4(CO)13]2- с Fe3(CO)12 вначале, главным образом, генерируются более тяжёлые ЖКАР Fe3(CO)11, Fe3(CO)12 и Fe4(CO)13. Соли [Fe3(CO)11]2- и [Fe4(CO)13]2- не реагируют с Fe(CO)5 при –80°C. Следы ЖКАР в этом случае можно детектировать лишь при +20°C через несколько десятков минут. Эти данные показывают, что при малых глубинах превращений в реакциях КФА и КЖ происходит одноэлектронный перенос с участием этих частиц.

Такой процесс можно рассматривать, как прямое одноэлектронное восстановление КЖ, что согласуется с высокими величинами восстановительных потенциалов КФА (см. рис. 5).

Рис. 5. Шкала редокс-потенциалов для КФА и ЖКАР и потенциалы восстановления Fe(CO)5 и Fe3(CO)12 в ТГФ.
Реакцию [Fe(CO)4]2- с Fe(CO)5, в которой происходит генерирование ЖКАР можно представить серией радикально-цепных превращений. При эквимолярном соотношении исходных соединений реакция приводит к образованию [Fe2(CO)8]2-.

При избытке Fe(CO)5 процесс идёт дальше с образованием на последних стадиях трёхъядерного КФА, уже не способного при нормальной температуре восстанавливать исходный пентакарбонил железа.

Радикально-цепной процесс, имеющий одноэлектронные редокс-стадии, заканчивается образованием диамагнитных комплексов. В закрытой системе, когда CO не выводится из сферы реакции, реализуется ситуация, при которой ЖКАР 2 – 5 находятся в равновесии.
Таким образом, КФА, являясь достаточно сильными восстановителями, способны участвовать в прямом одноэлектронном восстановлении различных субстратов, в том числе и КЖ, с образованием ЖКАР, которые претерпевают дальнейшие превращения в радикально-цепных процессах.
4. Окислительно-восстановительные реакции, в которых карбонилы железа являются восстановителями
Реакции диспропорционирования КЖ под действием оснований Льюиса протекают по схеме (уравнения 4, 5), включающей стадию активирующего комплексообразования и последующий одноэлектронный перенос с активированного донором электронов карбонильного комплекса на другую молекулу карбонила железа. Если в реакции присутствует третий компонент, способный восстанавливаться легче, чем КЖ, то возможно восстановление этого компонента, а не КЖ. Такая ситуация, представленная уравнениями (34) и (35), может реализовываться в случае соединений, потенциалы восстановления которых выше, чем у Fe(CO)5 (E red = -1.77 В). Это, например, нитрозосоединение t-BuNO (E red = -1.36 В), CCl4 (E red = - 0.78 В) и CHCl3 (E red = -1.67 В), а также сера (E red = -1.077 В).

Реакцию Fe(CO)5 с t-BuNO проводили в темноте непосредственно в резонаторе ЭПР-спектрометра. На рис. 6 представлена сильнопольная часть ЭПР-спектра.
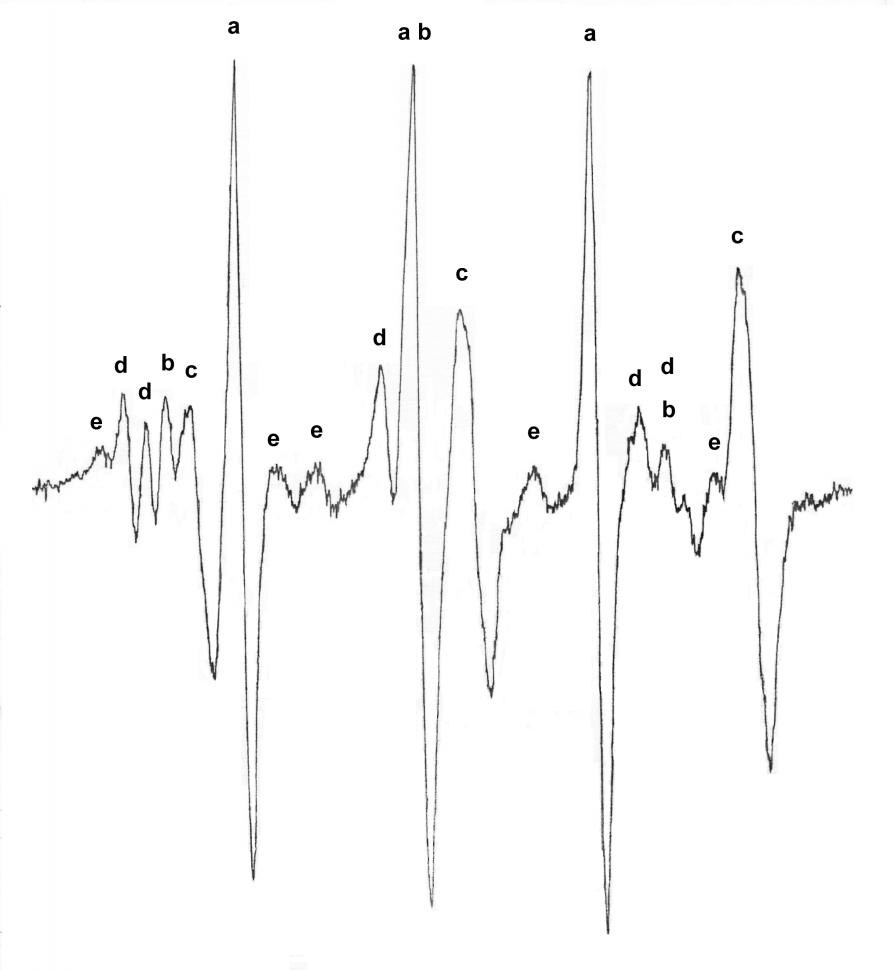 | Рис. 6. Сильнопольная часть ЭПР-спектра, наблюдаемого при взаимодействии Fe(CO)5 с t-BuNO в толуоле. После начала реакции температура понижена до – 80°C. Радикалы: a) tBuNO (g = 2.0060, aN = 10.7 мТл); b) tBu2NO (g = 2.0060, aN = 15.4 мТл); c) (CO)3(NO)FeN(O)-t-Bu (g = 2.0040, aN = 16.8 мТл, aN = 0.65 мТл); d) сигнал g = 2.0068 (триплет дублетов aN = 15.6 мТл, aH = 1.4 мТл), e) tBuNOH (g = 2.0060, aN = 13.1 мТл, aH = 10.7 мТл). |
Реакция имеет индукционный период, который обусловлен тем, что в качестве основания Льюиса (L в уравнениях 34 и 35) могут выступать либо основные центры на стеклянных стенках ампулы, либо слабоосновный мономер t-BuNO, который в растворе находится в равновесии с димером. Использование донорного растворителя (ТГФ) уменьшает индукционный период, а в случае основных реагентов, таких как ДМФА, ДМСО, [Et4N]Cl, PPNCl, индукционный период не наблюдается. На начальных стадиях после индукционного периода идёт интенсивное образование анион-радикала tBuNO и спинового аддукта железотрикарбонил-нитрозильного радикала. Далее идёт накопление ди-трет-бутилнитроксильного радикала. Анион-радикал t-BuNO, полученный по реакции (35) распадается по известному из литературы маршруту (уравнения 36, 37).

Образовавшийся анионный нуклеофил NO запускает уже известные схемы (38) и (39).

После окончания индукционного периода процесс протекает в радикально-цепном режиме, который обеспечивается образованием t-BuNO (уравнение 39) и его распадом (уравнение 36). При этом идёт накопление стабильных радикалов tBu2NO (уравнение 37) и (CO)3(NO)FeN(O)-t-Bu (уравнение 40). Если Fe(CO)5 берётся в избытке по отношению к t-BuNO, то после того, как израсходуется всё нитрозосоединение, осуществляется процесс восстановления образовавшегося tBu2NO (E red = -1.63 В)) до аниона гидроксиламина по схеме, аналогичной уравнениям (34), (35).
Таким образом, в реакции с участием t-BuNO, который способен восстанавливаться легче, чем Fe(CO)5, последний в составе активированного комплекса с основанием Льюиса выступает как одноэлектронный восстановитель. Образовавшийся анион-радикал t-BuNO определяет дальнейшую цепь превращений.
5. Роль железокарбонильных анион-радикалов в восстановительном карбонилировании нитро- и нитрозосоединений
Каталитические процессы карбонилирования с участием комплексов карбонилов железа включают два типа окислительно-восстановительных реакций. Это редокс-диспропорционирование КЖ под действием оснований Льюиса и окислительное присоединение кислот Бренстеда и Льюиса к КФА и ГКФА. В обоих случаях в итоге образуются ЖКАР Fe2(CO)8, Fe3(CO)11, Fe3(CO)12 и Fe4(CO)13, которые достаточно стабильны, и в процессе реакции их стационарные концентрации могут достигать высоких значений 10-5 – 10-2 моль·л -1. С другой стороны, ЖКАР – это координационно ненасыщенные и электронодефицитные системы, вследствие чего они имеют лабильную координационную сферу, в которой может происходить быстрый лигандный и электронный обмен. Реакции с их участием, как правило, имеют радикально-цепной характер, поэтому они должны подходить на роль катализаторов процессов карбонилирования органических соединений. Поэтому для исследования роли ЖКАР в таких процессах в качестве модельной была выбрана реакция восстановительного карбонилирования нитро- и нитрозосоединений. При прямом введении в реакцию ЖКАР из-за их высокой интенсивности не удаётся чётко идентифицировать другие сигналы радикальных комплексов, образующихся в системе. Поэтому для взаимодействия с нитро- и нитрозосоединениями мы использовали моно- и биядерные КФА. В этом случае скорость образования ЖКАР можно регулировать. Например, реакция Na2Fe(CO)4·1,5(diox) с t-BuNO2 в ТГФ протекает достаточно активно (рис. 7). При 80°C в первый момент времени образуются ЖКАР 2 – 4 (на рис. 7 обозначенные как 2, 3, 4) и сильно уширенный триплет (маркировка ‚) g = 2.0060, aN = 25.4 мТл, который, по лит. данным, отвечает tBuNO2. На более поздних стадиях процесса в спектре появляются ещё два сигнала от нитроксильных радикалов g = 2.0036, aN = 16.7 мТл (маркировка ) и g = 2.0053, aN = 16.7 мТл (маркировка ), а также сигналы от азотсодержащих радикалов 16 (g = 2.0267, aN = 1.8 мТл), 17 (g = 2.0251, aN = 2.3 мТл) и 18 (g = 2.0365).
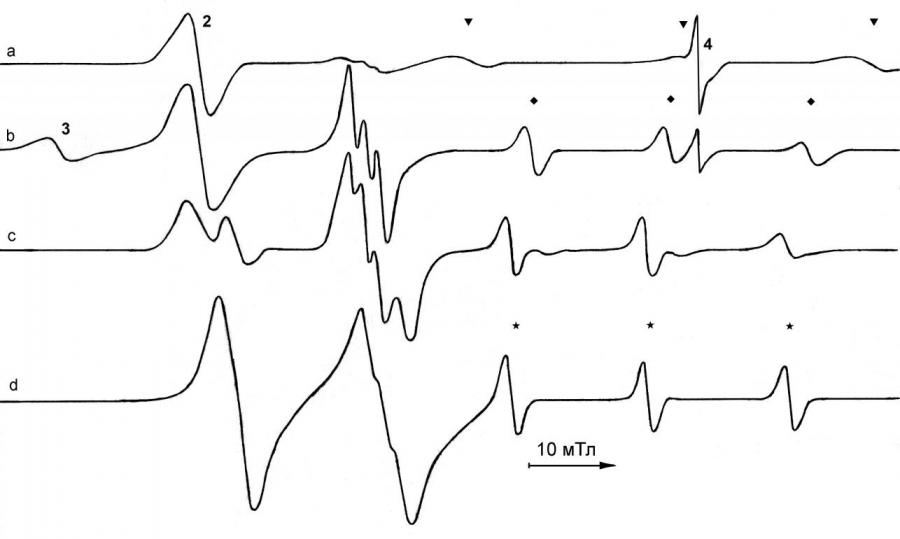  | Рис. 7. Спектры ЭПР, наблюдаемые при взаимодействии Na2Fe(CO)4·1.5(diox) c tBuNO2 в ТГФ. a) Через 1.5 мин после начала реакции, съёмка при 80°C; b) через 6 мин; c) после прогрева при 40°C через ~ 15 мин после начала реакции, съёмка при 60° C; d) через ~ 25 мин. |
В реакции [(PPN)2Fe2(CO)8] с t-BuNO2 в ТГФ (рис. 8) наблюдаются сигналы ЖКАР 2 – 4, радикала (16), а также очень слабые сигналы t-Bu2NO, помеченные на рис. 8 ‚.
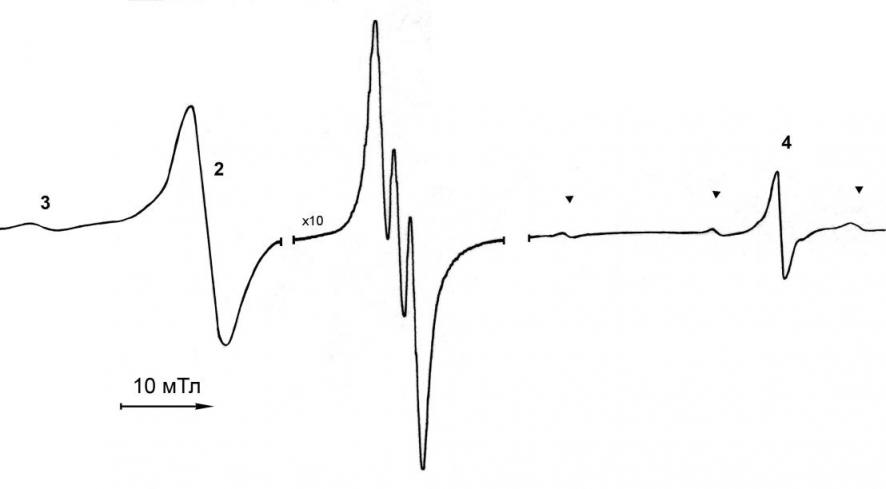 | Рис. 8. Спектр ЭПР, наблюдаемый при взаимодействии [(PPN)2Fe2(CO)8] с tBuNO2 в ТГФ при 60°C через 12 мин после начала реакции. |
Восстановление нитро- и нитрозотретбутана при действии КФА происходит с образованием как железокарбонильных, так и азоторганических анион-радикальных аналогов исходных соединений, что позволяет сделать вывод о механизме их взаимодействия. Первой стадией является прямой электронный перенос (уравнение 41).

Образовавшиеся в реакциях нитро- и нитрозотретбутана с КФА радикальные продукты можно условно разделить на четыре группы: В первую группу входят ЖКАР Fe2(CO)8, Fe3(CO)11, Fe3(CO)12. Вторую группу составляют известные нитроксильные радикалы с g = 2.0059 - 2.0063, образующиеся при восстановлении t-BuNO и t-BuNO2. Третья группа состоит из неизвестных ранее неустойчивых радикалов, g-факторы (2.0036 – 2.0062) и константы СТС (14.0 – 16.7 мТл) которых также находятся в нитроксильной области. Так как появление этих радикалов на первых стадиях реакций ЖКАР с t-BuNO не наблюдается, то, вероятно, они представляют собой спиновые аддукты не самих ЖКАР, а продуктов их превращения. Четвёртую группу образуют также неизвестные ранее азотсодержащие радикалы в области g = 2.025 - 2.036. Для выяснения строения радикалов третьей и четвёртой групп была проведена ещё одна серия реакций – восстановление на натриевом зеркале диамагнитных комплексов железа Fe3(CO)9(N-t-Bu)2 и Fe3(CO)10(N-t-Bu), которые образуются в реакциях КЖ с нитросоединениями. На рис. 9 показан спектр продуктов восстановления Fe3(CO)9(N-t-Bu)2. Сначала появляется сигнал квинтетной структуры от анион-радикала Fe3(CO)9(N-t-Bu)2 (g = 2.0135, aN = 1.8 мТл) (19), а затем - триплетные сигналы 16, 17 и сигналы от ЖКАР.
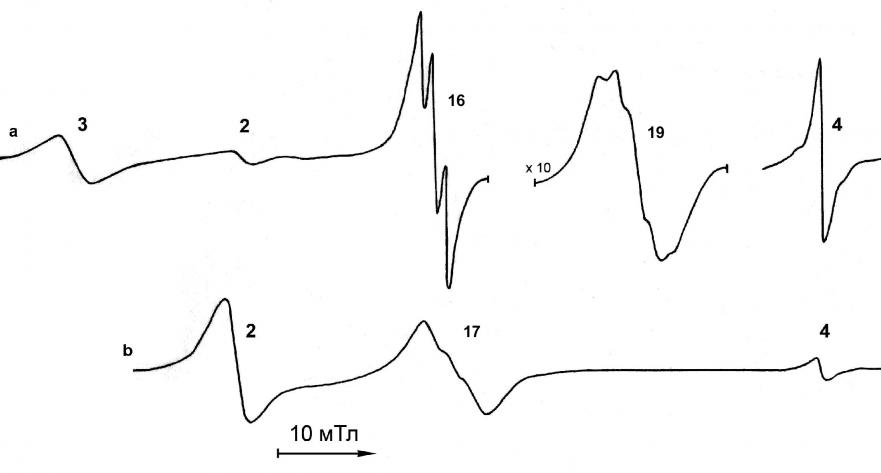 | Рис. 9. Спектры ЭПР, наблюдаемые при восстановлении Fe3(CO)9(N-t-Bu)2 на натриевом зеркале при -80 °C в ТГФ. a) Через 2 мин после начала реакции; b) через ~ 20 мин. |
При восстановлении Fe3(CO)10(N-t-Bu) при -80°C в ТГФ на первых стадиях в спектре ЭПР детектируются сигналы ЖКАР 3, 4, 2 и мультиплет, отвечающий радикалам 16 и 17. На более поздних стадиях появляется сигнал радикала 18.
Триплетная структура сигналов 16 и 17 свидетельствует о том, что в соответствующих радикалах неспаренный электрон взаимодействует с одним атомом азота. Их появление при восстановлении Fe3(CO)10(N-t-Bu) можно объяснить переходом 19 анион-радикала Fe3(CO)10(N-t-Bu) в 17 Fe3(CO)9(N-t-Bu). Образо-вание ЖКАР 2 – 4, не содержащих ни одного атома азота, при восстановлении Fe3(CO)9(N-t-Bu)2 и Fe3(CO)10(N-t-Bu) говорит о том, что происходит постепенное дезаминирование, состоящее в замене нитреновых лигандов молекулами CO в анион-радикалах Fe3(CO)9(N-t-Bu)2 и Fe3(CO)10(N-t-Bu). В реакциях восстановления Fe3(CO)9(N-t-Bu)2 и Fe3(CO)10(N-t-Bu) на натриевом зеркале с помощью масс-спектрометрии обнаружено образование t-BuNCO и мочевины (tBuNH)2CO. Сигналы 16 и 17, наряду с ЖКАР 2 – 4, образуются в реакции Fen(CO)m (n=1, m=5; n=2, m=9; n=3, m=12) и t-BuNCO при облучении светом лампы накаливания при -80°C в ТГФ. В этом случае контролируемый рост интенсивности сигнала Fe2(CO)8 позволяет детектировать радикалы 16 и 17.
Таким образом, возникновение радикалов 16 и 17 в реакциях карбонилферрат-анионов с третичными нитро- и нитрозобутаном, а также в реакциях карбонилов железа с третбутилизоцианатом указывает на то, что образование изоцианата происходит в результате карбонилирования нитреновых радикальных комплексов. Этим процессам отвечает схема 4:
 Cхема 4
Cхема 4
Восстановление осуществляется за счёт координированных CO-групп, которые при этом отщепляются в виде CO2. Если процесс идёт в каталитическом режиме при контролируемом отрицательном химическом потенциале среды (например, на натриевом зеркале), то происходит карбонилирование промежуточных нитреновых комплексов с образованием изоцианатов и исходных ЖКАР. При этом степень окисления атомов железа Fe(-I) в частицах – носителях каталитических функций остаётся неизменной. Каталитическая активность ЖКАР в реакциях восстановления нитросоединений впервые показана нами и, позднее, была подтверждена в исследованиях Ф.Рагаини и Г. Джеффроя (1995). Если реакцию проводить при стехиометрическом соотношении компонентов, то восстановление азотсодержащих лигандов может остановиться на стадии нитреновых структур. В отсутствие достаточного восстановительного потенциала среды и поступления дополнительного CO, восстановление азотсодержащих лигандов в промежуточных комплексах может идти за счёт окисления Fe(-I) до Fe(+I). Далее, теряя неспаренный электрон, анион-радикалы превращаются в известные диамагнитные комплексы с электронной конфигурацией железа d7, например Fe2(CO)6(NR)2, Fe3(CO)9(NR)2, Fe3(CO)10NR и др. Это приводит к выходу комплекса из каталитического цикла и делает невозможным дальнейшее образование изоцианатов (схема 4).
Таким образом, каталитический цикл восстановительного карбонилирования нитро- и нитрозосоединений с участием в качестве катализаторов ЖКАР включает превращения через 17 19 17 комплексы, и весь процесс протекает в радикально-цепном режиме.
6. Реакции солей феррициния с основаниями Льюиса
Исследование реакций натриевых солей азолов с гексафторфосфатом ферри-циния показало, что уже на первых минутах происходит быстрое изменение цвета реакционной смеси от синего до темно-красного. Хроматографическое разделение на Al2O3 реакционной смеси, образовавшейся через 5 - 75 мин при 20 40°C, показало (данные элементного анализа, масс-спектры), что среди продуктов присутствуют: ферроцен (~ 50%), N-ферроценилазол (5 40%), азол (10 40%), координационные полимеры (FeLx)n (~ 10%), небольшое количество циклопентадиеновых смол (~ 2%) и олигомеров тетрагидрофурана (следы). Выход N-ферроценилазолов падает в ряду бензотриазол > пиразол ~ 3,5-диметилпиразол > имидазол. Проведение реакций амидов и алкоголятов натрия с гексафторфосфатом феррициния показало, что амино- и алкоксиферроцены не образуются. При этом происходит частичное восстановление до ферроцена и деструкция с образованием циклопентадиеновых смол и неорганических солей железа.
ЭПР-спектроскопическое изучение реакций гексафторфосфата феррициния с натриевыми солями азолов (пиразола, 3,5-диметилпиразола, бензотриазола) показало, что независимо от природы исходного азолид-аниона, вначале при -80°C в спектре появляется очень широкий сигнал (H ~250 мТл) с g-фактором, близким к электронному (g = 2.0026 2.0028), который не соответствует ни катион-радикалу феррициния (может появляться только при очень низких температурах g = 4.40 и g = 1.39), ни азолильным радикалам, имеющим мультиплетную структуру. Не соответствует он и неорганическому Fe(II) (g = 3.30 3.43 и g = 6.58 6.86). Вероятно, этот сигнал может принадлежать системам с переносом заряда [Fс•+ Az–] (Az–- азолид-ион). При повышении температуры этот сигнал быстро уменьшается с одновременным появлением в ЭПР-спектре триплета триплетов спиновых аддуктов азолильных радикалов (в присутствии спиновой ловушки t-BuNO). Совокупность химических и спектральных данных позволяет сделать заключение, что в этих реакциях реализуется схема прямого восстановления катион-радикала феррициния до ферроцена анионами азолов (уравнение (42)). Азолильные радикалы обладают достаточно высокой реакционной способностью и взаимодействуют с другим катион-радикалом феррициния по месту локализации неспаренного электрона, то есть по атому железа. Основным электронным состоянием катион-радикала феррициния является 2E2g с конфигурацией (a21g e32g). Термическая заселённость первого возбуждённого состояния 2A1g (a11g e42g) при комнатной температуре значительна, так как электронный переход 2E2g 2A1g лежит в области около 200 см-1, что соответствует в этих условиях kT. В зависимости от того, в каком электронном состоянии находится катион феррициния, осуществляется либо рикошетная атака в циклопентадиенильное кольцо, либо образование координационной связи Fe—N. Первое приводит к замещению водорода и образованию N-ферроценилазолов (уравнения 43 и 44), второе к распаду ферроценового ядра и координационным полимерам (FeAzx)n. В работе (В.А. Нефёдов, 1976) ключевой стадией процесса также считается взаимодействие катион-радикала феррициния с радикалом, полученным при окислении основания (уравнение 43).

В тех случаях, когда основания Льюиса, взятые в анионной форме, не окисляются до радикалов, возможно их комплексообразование с катион-радикалом феррициния как это происходит, например, с гидроксильным анионом (уравнение 45, где L– = OH–). При этом образуется гидроксид феррициния, который удаётся выделить в виде жёлтого аморфного порошка (Абакумова, 1978). Его взаимодействие с солью феррициния приводит к быстрой реакции (46) с выделением ферроцена.
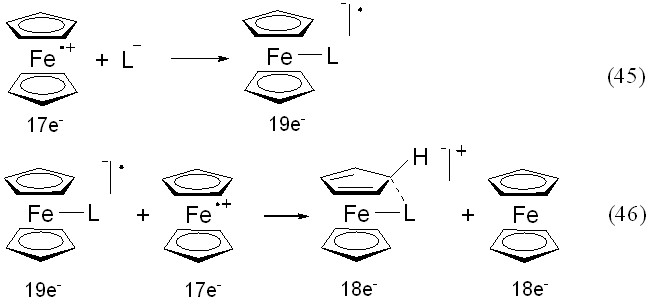
Образующийся в реакции (46) 18е катион при взаимодействии с основа-ниями Льюиса распадается до неорганических комплексов Fe(II), которые во вторичных процессах окисляются исходным катионом феррициния до комплексов Fe(III): со щелочами образуется Fe(OH)3, c NH4SCN - анион [Fe(SCN)6]3-; с Cl– и Br– - анионы [FeCl4]– и [FeBr4]– соответственно. Приведенный в уравнениях (45), (46) механизм соответствует концепции предварительного «активирующего комплексообразования» с последующей одноэлектронной редокс-стадией.
Таким образом, при взаимодействии катион-радикала феррициния с основаниями Льюиса замещение в циклопентадиенильное кольцо с образованием ферроценовых производных происходит только в тех случаях, когда в процессе реакции из оснований Льюиса образуются радикалы, которые затем непосредственно реагируют с катион-радикалом феррициния. В том случае, когда окисление основания Льюиса не происходит, восстановление катион-радикала феррициния протекает через стадию образования комплекса катион-радикала феррициния с основанием с последующей передачей электрона на другой катион-радикал феррициния. При этом часть катион-радикала феррициния восстанавливается до ферроцена, а часть при дальнейшем взаимодействии с основанием Льюиса превращается в координационное соединение железа.
7. Термическая диссоциация карбонилов железа в условиях роста нитевидных кристаллов железа[1]
Исследования термической диссоциации Fe(CO)5, Fe3(CO)12 и Fe5С(СО)15 в газовой фазе показали, что в результате на подложке образуются нитевидные монокристаллы (НК) -железа (рис. 10).
 | Рис. 10. Прямые НК -Fe с разными направлениями роста, полученные при температуре подложки 1000°С и давлении Fe(CO)5 1.510-1 Торр. |
Было установлено, что рост НК осуществляется в две стадии: сначала формируется подслой, а уже затем образуются НК. Они начинают расти только на дискретных, куполообразных (полусфероидных) структурах подслоя размером около 1 мкм - своеобразных зародышах. Рост НК осуществляется в довольно узком интервале температур (примерно 50°C), который ограничен как сверху, так и снизу, т.е. зависимость роста НК от температуры имеет пороговый характер. Аналогично температурному, существует узкий диапазон давлений Fe(CO)5 с нижней и верхней границами, в котором происходит рост НК. Максимальная длина НК -Fe и их плотность при получении из Fe(CO)5 приходится на диапазон температур 1000 – 1050°С и давлений 1.510-1 510-1.
Проведено ЭПР-спектроскопическое исследование реакции термодиссоциации карбонилов железа в условиях регистрации спектров ЭПР, близких к условиям получения НК. Было зарегистрировано семейство сигналов со значениями g-факторов в диапазоне 1.9 – 2.1, которые отвечают ЖКАР с различным числом атомов железа от Fe1 до Fe4. Все зарегистрированные при -90°С сигналы можно разделить на две группы: с изотропными g-факторами (Fe(CO)4, g = 2.0485; Fe2(CO)8, g = 2.0394; Fe3(CO)11, g = 2.0493; Fe3(CO)12, g = 2.0018; Fe4(CO)13, g = 2.0134) и анизотропными значениями g-факторов (Fe(CO)4, g1 = 2.0710, g2 = 2.0710, g3 = 2.0038; Fe2(CO)8, g1 = 2.0566, g2 = 2.0523, g3 = 2.0090; Fe3(CO)11, g1 = 2.0927, g2 = 2.0314, g3 = 2.0247). Это вызвано тем, насколько жестко анион-радикалы связаны с поверхностью. Жестко-связанные ЖКАР (хемосорбированные) дают сигналы с анизотропными, а свободные ЖКАР (физически адсорбированные) - с изотропными значениями g-фактора. Увеличение времени нагрева образца не приводило к заметным изменениям спектров. Единственным ограничением служил рост толщины покрытия, что делало невозможным измерение спектров ЭПР из-за образования магнитной фазы, приводящей к падению добротности резонатора. Повышение температуры приводило не к ослаблению сигналов, а к их уширению, что связано с протеканием реакций электронного обмена в адсорбционном слое.
На основании полученных данных была предложена модель цепного ион-радикального механизма термодиссоциации карбонилов металлов с зарождением цепи по схеме активирующего комплексообразования, которая включает все характерные для цепных процессов этапы: зарождение цепи и возникновение активных железо-карбонильных анион-радикалов; продолжение цепи; укрупнение и взаимопревращения ЖКАР; формирование структуры кристаллов -железа и обрыв цепи; гибель ион-радикальных центров и прекращение быстрого роста НК в длину.
Этап зарождения цепи начинается с адсорбции налетающей из газовой фазы молекулы ПКЖ на адсорбционном донорном центре (Sд) исходной подложки. Такие центры либо обусловлены примесями, либо образуются в результате атмосферного воздействия на подложку. После адсорбции происходит зарождение ион-радикального центра, что определяет радикальную природу процесса в целом. Можно выделить два типа реакций зарождения: на примесных группах атомов донорной природы исходной подложки и на поверхности сферических зародышей уже сформировавшегося на первичной подложке металлического подслоя, причем в обоих случаях ключевая роль принадлежит одноэлектронному переносу. В случае инициирования на исходных подложках происходит окислительно-восстановительное диспропорционирование молекул Fe(CO)5 за счет электронов донорных центров подложки, в результате чего на поверхности возникают реакционноспособные частицы с открытой электронной оболочкой (схема 5).
 Схема 5
Схема 5
Этот низкоэнергетический процесс протекает при относительно низких температурах, и металлическое покрытие-подслой, на котором в предложенной схеме происходит зарождение цепи, образуется уже при 250 °С.
В случае зарождения цепи на поверхности сферических зародышей уже сформировавшегося на первичной подложке металлического подслоя донором электронов является поверхностная группа атомов железа. Для того чтобы осуществить перенос электрона к молекуле адсорбата, необходимо затратить энергию по преодолению относительно большой работы выхода электрона из железа (1.5 еV), что обуславливает сильную зависимость роста НК от температуры и существование нижнего температурного предела образования НК, который намного выше температурного предела начала образования покрытий, а также зависимость от размера зародыша. При повышении температуры происходит заселение высоко лежащих вакантных электронных уровней в зоне проводимости, что и делает возможным электронный перенос с металлического зародыша на молекулу Fe(CO)5 (схема 6).

Схема 6
На этапе продолжения цепи (схема 7), образовавшиеся на предыдущей стадии адсорбированные анион-радикалы быстро реагируют с нейтральными молекулами карбонила железа, в результате чего образуется семейство железокарбонильных анион-радикальных кластеров с различным числом атомов металла
 и т.д. Схема 7
и т.д. Схема 7
Последний этап процесса термораспада - обрыв цепи, приводящий к прекращению роста НК, осуществляется путем возврата свободного электрона ЖКAP в зону проводимости и сопровождается образованием нейтрального адсорбционного комплекса (схема 8). Гибель анион-радикальных адсорбционных комплексов, ведущих цепь, может произойти либо при понижении температуры, либо при введении добавок, вызывающих «рекомбинацию» ЖКАР.

Схема 8
Таким образом, предложенная нами модель позволяет непротиворечиво объяснить всю совокупность экспериментальных фактов, касающихся характерных особенностей состава и морфологии НК и кинетики их роста. Выявленные закономерности для карбонилов железа показывают общность ион-радикальных процессов кластеризации, протекающих в газовой, жидкой фазе и в адсорбционном слое. Они хорошо согласуется с моделями цепных ион-радикальных процессов, инициированных активирующим комплексообразованием, предложенными для жидкой и газовой фаз.
Выводы
1. Совокупностью кинетических и синтетических методов, в сочетании с данными ЭПР-спектроскопии, изучены реакции карбонилов железа с основаниями Льюиса, а также карбонилферрат-анионов и гидридокарбонилферрат-анионов с кислотами Льюиса (Бренстеда) и установлено, что на первых стадиях этих взаимо-действий происходит одноэлектронное редокс-инициирование с предварительным активирующим комплексообразованием, в результате чего образуются радикальные и ион-радикальные комплексы железа, которые инициируют развитие двух цепей превращений, обусловленных реакциями замещения в координационной сфере металла. Показано, что карбонилферрат-анионы, являясь достаточно сильными восстановителями, способны участвовать в прямом одноэлектронном восстановлении карбонилов железа, с образованием железокарбонильных анион-радикалов, которые претерпевают дальнейшие превращения в радикально-цепных процессах.
2. Предложена схема каталитического процесса восстановительного карбонилирования нитро- и нитрозосоединений в присутствии карбонилов железа, в котором роль каталитически активных частиц выполняют железокарбонильные анион-радикалы Fe2(CO)8, Fe3(CO)11 и Fe4(CO)13. Показано, что этот каталитический цикл включает превращения через 17 19 17 комплексы, и весь процесс протекает в радикально-цепном режиме. Выявленные закономерности позволяют использовать их для поиска на основе карбонилов металлов эффективных катализаторов процессов карбонилирования при контролируемом электрохимическом потенциале среды.
3. Установлено, что реакции пентакарбонила железа с реагентами (2-метил-2-нитрозопропан, сера), которые способны восстанавливаться легче, чем пентакарбонил железа, протекают через редокс-стадии, в которых пентакарбонил железа в составе активированного комплекса с основанием Льюиса выступает как одноэлектронный восстановитель, а реагент как окислитель. Дальнейшие превращения определяются образовавшимися анион-радикалами реагентов.
4. Впервые получены и охарактеризованы новые производные ферроцена - N-ферроценилазолы и показано, что их образование осуществляется путём присоединения к катион-радикалу феррициния азолильных радикалов, которые возникают при непосредственном окислении азолид-анионов солями феррициния. В этой реакции реализуются два типа редокс-процессов — одноэлектронный перенос и редокс-инициирование с предварительным активирующим комплексообразованием.
5. Разработан метод термической диссоциации карбонилов железа в газовой фазе, с помощью которого впервые получены нитевидные монокристаллы -железа. Установлены основные факторы (давление и температура), определяющие скорость роста и свойства кристаллов и найдены оптимальные условия их получения. Методом ЭПР обнаружено образование железокарбонильных анион-радикалов в условиях газофазной термической диссоциации карбонилов железа и показано, что этот процесс осуществляется по цепному ион-радикальному механизму в адсорбционном слое через стадию активирующего комплексообразования с металлической поверхностью.
6. Предложен общий подход к описанию взаимодействий карбонилов металлов с основаниями Льюиса в рамках схемы редокс-диспропорционирования, включающей стадию активирующего комплексообразования с последующим одноэлектронным переносом с активированного донором электронов карбонильного комплекса на другую молекулу карбонила металла. Превращения образовавшихся радикальных и ион-радикальных комплексов могут осуществляться по двум направлениям: диспропорционирование с образованием стабильных, катионных комплексов металлов, содержащих в координационной сфере основания Льюиса, и карбонилметаллат-анионов (или гидридокарбонилметаллат-анионов); радикально-цепные процессы катализа, индуцируемого электронным переносом с образованием диамагнитных производных карбонильных комплексов металлов.
Основное содержание работы изложено в следующих публикациях:
- Ю. А. Белоусов. Радикальная химия карбонилов железа. Успехи химии, 76, 46-65 (2007)
- В. Н. Бабин, Ю. А. Белоусов, И. В. Добрякова, Ю. С. Некрасов, В. Г. Сыркин, А. А. Уэльский. Термическая диссоциация карбонилов железа в условиях роста нитевидных кристаллов железа. Изв. АН, Сер. хим., №9, 1826-1836 (2004)
- Yu. A. Belousov, E. F. Brin. Investigation of the kinetics of the chain-radical process in the system iron carbonyl Lewis base. Polyhedron, 20, 2765-2769 (2001)
- Yu. A. Belousov, T. A. Belousova. The formation of iron carbonyl radical anion in the reaction of iron carbonyls with trimethylamine N-oxide. Polyhedron, 18, 2605-2608 (1999)
- Ю. А. Белоусов. Взаимодействие карбонилферрат- и гидридокарбонилферрат-анионов с кислотами Бренстеда и Льюиса. Изв. АН. Сер. хим.,1048-1054 (1997)
- Ю. А. Белоусов, Т. А. Белоусова. Взаимодействие карбонилов железа с основаниями Льюиса. Изв. АН. Сер. хим.,248-249 (1996)
- Yu. A. Belousov, T. A. Kolosova, V. N. Babin. Interaction of iron carbonyls with Lewis bases. Polyhedron, 8, 603-611 (1989)
- Yu. A. Belousov, T. A. Kolosova. ESR Study on the reaction of iron carbonyls with nitro and nitrosoparaffines. A mechanism of the reductive carbonylation of nitro compounds. Polyhedron, 6, 1959-1970 (1987)
- Ю. А. Белоусов, Т. А. Колосова, Е. А. Викторова, В. А. Сергеев, В. Н. Бабин. Изучение взаимодействия карбонилов железа с алкилтиолятами натрия методом ЭПР. Изв. АН.СССР, Сер. хим.,1652-1654 (1987)
- V. N. Babin, Yu. A. Belousov, V. V. Gumenyuk, R. M. Salimov, R. B. Materikova, N. S. Kochetkova. Reactions of anionic nitrogen heterocycles with iron carbonyls. J. Organomet. Chem., 241, C41-C44 (1983)
- Ю. А. Белоусов, В. В. Гуменюк, В. Н. Бабин, Н. С. Кочеткова, С. П. Солодовников. Комплексы карбонилов металлов с азолами. Исследование химического поведения солей ди- и триазолов в реакции с -аллилтрикарбонилиодидом железа. Корд. химия, 9, 819-824 (1983)
- V. V. Gumenyuk, V. N. Babin, Yu. A. Belousov, N. S. Kochetkova, I. V. Dobryakova. Reactions of Diazoles with Manganese and Iron Carbonyls. Polyhedron, 3, 707-711 (1984)
- Ю. А. Белоусов, Т. А. Колосова, В. Н. Бабин, В. А. Сергеев, Е. А. Викторова. Способ получения бис(алкилтиотрикарбонилжелеза). Авторское свид. СССР № 1384593, Бюлл. изобр., № 12 (1988)
- В. Г. Сыркин, А. А. Уэльский, И. В. Добрякова, А. С. Рыжов, И. С. Толмасский, М. А. Хацернов, В. Н. Бабин, Ю. А. Белоусов, Т. Ю. Сорокина. Способ получения нитевидных кристаллов («усов») из железа, хрома, молибдена и вольфрама. Авторское свид. СССР № 1450399, н/п., (1989)
- V. N. Babin, Yu. A. Belousov, I. R. Lyatifov, R. B. Materikova, V. V. Gumenyuk. Azole Anions as One-Electron Reducers of Ferricinium Salts. J. Organomet. Chem., 214, C11-C12 (1981)
- V. N. Babin, Yu. A. Belousov, V. V. Gumenyuk, R. B. Materikova, R. M. Salimov, N. S. Kochetkova. Direct Heteroarylation of ferricinium Salts. J. Organomet. Chem., 241, C41-C44 (1983)
- V. N. Babin, V. V. Gumenyuk, S. P. Solodovnikov, Yu. A. Belousov, A spin trap investigation of azolyl radicals. Z. Naturforsch., 36b, 400-401 (1981)
- Ю. А. Белоусов, Т. А. Белоусова. Радикальное восстановление нитрозосоединений пентакарбонилом железа. International Symposium on Advanced Science in Organic Chemistry, Sydak, Crimea, 2006, C-016
- Yu.A. Belousov, T.A. Belousova, Radical Chemistry of Iron Carbonyls. International Conference Dedicated to 50th Anniversary of A.N. Nesmeyanov Institute of Organoelement Compounds “Modern Trends in Organoelement Compounds and Polymer Chemistry”. Moscow, May 30-June 4, 2004, Book of Abstracts, p. 119.
- Ю.А. Белоусов, Т.А. Белоусова. Радикальная химия карбонилов железа. Сб. материалов второй региональной научной конференции по органической химии, Липецк, 2000 , с. 52
- Ю.А. Белоусов, Т.А. Белоусова. Радикальная химия карбонилов железа. Тезисы докладов VI Всероссийской конференции по металлоорганической химии, Нижний Новгород,1995, с. 212
- Yu. A. Belousov. Activating Complexing Induced Radical Chain Reactions between Iron Carbonyls and Various Nucleophilic and Electrophilic Reagents. Proccedings of the X-th FECHEM Conference on Organometallic Chemistry. Agia Pelagia, Crete, Greece, 1993. р. 108
- Yu. A. Belousov, T. A. Kolosova, V. N. Babin. Interaction of iron carbonyls with Lewis bases. Proccedings of the VIII-th FECHEM Conference on Organometallic Chemistry, Hungary, 1989, P-68
- Ю. А. Белоусов, Т. А. Колосова, В. Н. Бабин. Взаимодействие карбонилов железа с основаниями Льюиса. Тезисы докладов IV Всесоюзной конференции по металлоорганической химии. Ч. 3, Казань,1988, с. 39
- В. Н. Бабин, Ю. А. Белоусов, В. В. Гуменюк, Р. М. Салимов, Л. И. Попова, В. И. Боев, Р. Б. Материкова. Азолы в реакциях с металлоорганическими соединениями переходных металлов. IV Всесоюзная конференция по химии азотсодержащих гетероциклических соединений, Новосибирск, 1987, с. 71
- Yu. A. Belousov, T. A. Kolosova, V. N. Babin. ESR-Studies on the Role of Iron Carbonyl Ion-Radicals for the Synthesis of Organo-Nitrogen Compounds. VI International Conference on Organic Synthesis, IUPAC, Moscow, 1986, р. 231
- N. S. Kochetkova, L. V. Popova, R. M. Salimov, Yu. A. Belousov, V. I. Boev, R. B. Materikova, V. N. Babin. Ferrocen derivatives - in synthesis of substituted nitrogen heterocycles. VI International Conference on Organic Synthesis, IUPAC, Moscow, 1986, р. 204
- Ю. А. Белоусов, Е. И. Мысов. ЭПР-исследование взаимодействия карбонилов железа с нитрозо- и нитросоединениями. IV Международный симпозиум по гомогенному катализу, Ленинград, 1984, т. IV, с. 30
- Yu. A. Belousov, V. N. Babin, V. V. Gumenyuk, R. M. Salimov, N. S. Kochetkova, R. B. Materikova. Reactions of five-membered nitrogen heterocycles with organometallic compounds of iron. Proccedings of the IV-th FECHEM Conference on Organometallic Chemistry, Liblice, Czechoslovakia, 1982, A-4
- В. Н. Бабин, Ю. А. Белоусов, В. В. Гуменюк, Р. Б. Материкова, Р. М. Салимов, Н. С. Кочеткова. Анионы азолов как восстановители металлоорганических соединений железа. Реакционная способность азолильных радикалов. Тезисы докладов II Всесоюзной конференции по металлоорганической химии, Горький,1982, с. 244-245
- А. Н. Несмеянов, Ю. А. Белоусов, В. В. Гуменюк, В. Н. Бабин, Н. С. Кочеткова. Комплексы карбонилов металлов с азолами. Исследование химического поведения солей ди- и триазолов в реакции с -аллилтрикарбонилиодидом железа. Тезисы докладов I Всесоюзной конференции по металлоорганической химии, Москва,1979, ч.I, № 165
- I. V. Dobryakova, V. G. Syrkin, V. N. Babin, Yu. A. Belousov, A. A. Uelskii. Cluster mechanism of the thermal dissociation of V-VIII group metal carbonyls. XXVIII International Conference on Coordination Chemistry, Gera, GDR, 1990, 5-24
- V. G. Syrkin, I. V. Dobryakova, V. N. Babin, Yu. A. Belousov, A. A. Uelskii. Catalytic function of paramagnetic iron carbonyl clasters in Fe(CO)5 thermal dissociation. Proccedings of the VIII-th FECHEM Conference on Organometallic Chemistry, Hungary, 1989, р. 52
- И. В. Добрякова, В. Г. Сыркин, А. А. Уэльский, В. Н. Бабин, Ю. А. Белоусов, А. И. Сыроваткин. Исследование некоторых свойств металлических нитевидных кристаллов железа, хрома, молибдена и вльфрама. Тезисы докладов I Украинской республиканской конференции «Газофазное получение новых функциональных материалов и плёнок», Вып. 1, Ужгород, 1989, с. 7-8
- В. Г. Сыркин, И. В. Добрякова, В. Н. Бабин, Ю. А. Белоусов, А. А. Уэльский, А. А. Скачков, С. В. Шелудякова. Влияние реакции Белла-Будуара на формирование карбидных фаз в процессе термораспада пентакарбонила железа. Тезисы докладов IV Всесоюзной конференции по металлоорганической химии. Ч. 2, Казань,1988, с. 70
- В. Г. Сыркин, В. Н. Бабин, А. А. Уэльский, Ю. А. Белоусов. Роль парамагнитных кластеров в процессах термической диссоциации карбонилов металлов. Тезисы докладов IV Европейской конференции по металлоорганической химии, Рига, 1985, с. 153
- И. В. Добрякова, А. А. Уэльский, В. Г. Сыркин, М. А. Хацернов, В. Н. Бабин, Ю. А. Белоусов. Получение, свойства и применение нитевидных монокристаллов тугоплавких металлов из карбонильной газовой фазы. XII Всесоюзное совещание. Получение, структура, физические свойства и применение высокочистых и монокристаллических тугоплавких и редких металлов, Суздаль, 1987, с. 6
[1] Исследования по термодиссоциации КЖ, описанные в этом разделе, были выполнены с сотрудниками лаборатории карбонильных материалов ГНИИХТЕОС: В. Г. Сыркиным, А. А. Уэльским, И. В. Добряковой. Электронно-спектроскопические исследования выполнены М. А. Хацерновым. Автор приносит им глубокую благодарность за помощь и сотрудничество.