

Взаимодействие органических гетерофункциональных производных кремния и германия с 1-алкинами
На правах рукописи
Коншин Валерий Викторович
ВЗАИМОДЕЙСТВИЕ ОРГАНИЧЕСКИХ ГЕТЕРОФУНКЦИОНАЛЬНЫХ ПРОИЗВОДНЫХ
КРЕМНИЯ И ГЕРМАНИЯ С 1-АЛКИНАМИ
Специальность: 02.00.03 – Органическая химия
АВТОРЕФЕРАТ
диссертации на соискание ученой степени
кандидата химических наук
Краснодар-2009
Работа выполнена в Кубанском государственном университете
Научный руководитель: кандидат химических наук,
старший научный сотрудник
Андреев Алексей Алексеевич
Официальные оппоненты: доктор химических наук, профессор
Косулина Татьяна Петровна
кандидат химических наук,
старший научный сотрудник
Журавлев Сергей Васильевич
Ведущая организация: Иркутский институт химии
им. А. Е. Фаворского
Сибирского отделения Российской академии наук
Защита состоится «22» декабря 2009 г. в ауд. 94 в 14-40 часов на заседании диссертационного совета Д 212.100.01 в Кубанском государственном технологическом университете по адресу:
г. Краснодар, ул. Красная, 135 (адрес для переписки 350072, г. Краснодар, ул. Московская, 2, корпус А)
С диссертацией можно ознакомиться в библиотеке Кубанского государственного технологического университета по адресу:
350072, г. Краснодар, ул. Московская, 2, корпус А
Автореферат разослан « 20 » ноября 2009 г.
Ученый секретарь
диссертационного совета
кандидат химических наук,
![]()
доцент Кожина Н.Д.
ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ
Актуальность темы. Значение использования кремнийзамещенных ацетиленовых соединений в такой области химической науки и практики, как тонкий органический синтез, трудно переоценить. Замещение подвижного атома водорода 1-алкинов на триорганилсилильную группу широко используется как прием химической защиты концевой ацетиленовой связи. Введение атома кремния к углероду тройной связи 1-алкинов изменяет реакционную способность тройной связи, повышает регио- и стереоселективность многих реакций присоединения по кратной связи. В последние годы силилацетилены стали использоваться в реакциях сочетания с функциональными органическими молекулами, приводящих к образованию новых углерод-углеродных связей. Таким образом, кремнийацетиленовые соединения превратились в эффективный инструмент конструирования сложных органических молекул.
Ацетиленовые производные кремния и германия в последнее время находят и практическое использование в качестве материалов полупроводниковой техники, исходных мономеров для создания органических полупроводников и проводящих материалов. Однако, для получения силил- и гермилалкинов в настоящее время используются те же реакции, с помощью которых в своё время были открыты первые представители ацетиленовых соединений элементов IV-a группы. Основанные на использовании ацетиленидов активных металлов, приемы создания кремнийацетиленовых связей делают силилацетилены труднодоступными соединениями, затрудняют освоение масштабного промышленного синтеза этих веществ. В плане этого, представляется высоко актуальным поиск новых путей прямого синтеза ацетиленовых соединений кремния и германия, основанных на оригинальных подходах к методам создания связи элемент-углерод.
Работа выполнялась в соответствии с планом научной работы кафедры органической химии и технологий ГОУ ВПО «Кубанский государственный университет», а также при финансовой поддержке РФФИ - проект 06-03-96667-р_юг_а «Взаимодействие органических гетерофункциональных производных кремния и германия с 1-алкинами».
Цель работы. Разработка новых методов синтеза ацетиленовых соединений кремния и германия, основанных на реакциях прямого силилилрования/гермилирования 1-алкинов под действием функциональных силанов и германов.
Выполнение поставленной цели потребовало решения следующих задач:
-изучение реакционной способности связей кремния и германия с гетероатомами в реакциях с 1-алкинами в присутствии кислот Льюиса и органических оснований;
-определение реакционной способности различных терминальных ацетиленовых соединений, в том числе и функционально замещенных, в процессах взаимодействия с галоген- и аминосиланами и германами;
-разработка препаративных методик получения триалкилсилилацетиленов, диалкилдиалкинилсиланов, алкилтриалкинилсиланов, три- и тетраалкинилгерманов из соответствующих галоген- и аминопроизводных кремния и германия;
-определение реакционной способности гетероатомных производных триметилкремния по отношению к тетрафенилэтинилаланату лития;
-обоснование химизма исследуемых реакций, выявления их основных закономерностей и синтетических возможностей.
Научная новизна. Впервые проведены систематические исследования взаимодействия 1-алкинов с азот-, кислород- и галогенсодержащими силанами и германами, в том числе и в условиях активации реакций кислотами Льюиса. Обнаружена способность диалкиламиноорганосиланов к эффективному взаимодействию с 1-алкинами в присутствии галогенидов цинка, кадмия и индия, приводящему к образованию силилацетиленов с высокой конверсией исходных веществ. На основе изучения реакционной способности различных 1-алкинов в данной реакции сделаны выводы о химизме реакции, получены данные, свидетельствующие об электрофильном характере процесса силилирования 1-алкинов при взаимодействии их с диалкиламиноорганосиланами в присутствии галогенидов цинка.
Обнаружена ранее неизвестная способность три- и тетрадиалкиламиногерманов к взаимодействию с 1-алкинами в присутствии галогенидов цинка с образованием соответствующих три- и тетраалкинилидов германия.
Определены условия взаимодействия различных галогенсиланов, галогенгерманов, О-силилкарбаматов с 1-алкинами, способствующие протеканию реакций силилирования ацетиленовых субстратов. Высказаны предположения о возможных механизмах изученных реакций.
Исследована реакционная способность кремнийорганических соединений, содержащих связь Si-N, Si-O, Si-Hal, по отношению к тетрафенилэтинилаланату лития.
Практическая значимость. Разработаны препаративные методы синтеза широкого ряда кремний- и германийацетиленовых соединений, таких как триалкилалкинилсиланы, диалкилдиалкинилсиланы, алкилтриалкинилсиланы, три- и тетраалкинилгерманы. Новые способы получения этих соединений выгодно отличаются от ранее используемых простотой исполнения, доступностью исходных веществ, одностадийностью. Важным преимуществом предлагаемых методов синтеза является возможность получения силилированных алкинов и алкинилгерманов, содержащих реакционноспособные атомы и функциональные группы. Данные методы синтеза могут быть легко осуществлены в промышленных масштабах и позволяют синтезировать многие, ранее труднодоступные, ацетиленовые соединения кремния и германия для использования в качестве эффективных реагентов органического синтеза.
Апробация работы. Основные результаты работы доложены на: ХIХ Международной научно-технической конференции РЕАКТИВ 2006 "Химические реактивы и процессы малотоннажной химии" (Уфа, 2006 XVII, XVIII Менделеевском съезде по общей и прикладной химии (Казань,2003; Москва, 2007), VII, Х Молодежной конференции по органической химии (Уфа, 2007), XVI, XVII Российской молодежной научной конференции «Проблемы теоретической и экспериментальной химии», (Екатеринбург, 2006, 2007), 10 Всероссийской конференции «Кремнийорганические соединения: синтез, свойства, применение», (Москва, 2005).
Публикации. По материалам диссертации опубликованы 2 статьи в журналах, рекомендованных перечнем ВАК, 2 статьи в сборниках научных трудов, 13 тезисов докладов международных и всероссийских конференций. Получен патент РФ.
Структура и объем работы. Работа состоит из введения, обзора литературы, экспериментальной части, обсуждения результатов, выводов, списка цитируемой литературы. Диссертационная работа изложена на 170 страницах, содержит 23 таблицы, 5 рисунков и библиографию из 265 наименований.
ОСНОВНОЕ СОДЕРЖАНИЕ РАБОТЫ
Во введении раскрыта актуальность темы диссертационной работы, обоснован выбор объектов исследования, определены цели и задачи исследования.
В литературном обзоре проанализированы данные о методах синтеза алкинилсиланов, опубликованные в научной периодике в последние 30 лет. Особое внимание уделено прямым методам силилирования 1-алкинов, обсуждены основные достоинства и недостатки этих методов.
Во второй главе изложены результаты исследования с их обсуждением.
В третьей главе представлены данные об используемых методах и приборах, описаны методики эксперимента, приведены физико-химические константы и спектральные характеристики синтезированных соединений.
1 Синтез ацетиленовых производных германия
1.1 Взаимодействие тетрахлорида германия и фенилтрихлоргермана с 1-алкинами
Для увеличения глубины алкинилирования GeCl4 по известной реакции последнего с 1-алкинами в присутствии третичных аминов, нами было предложено проводить данное взаимодействие в присутствии кислоты Льюиса, способной образовывать с галогенид-анионом комплексный анион, не способный играть роль нуклеофила по отношению к германию. Данная кислота Льюиса не должна обладать способностью вызывать полимеризацию 1-алкина, с одной стороны, а также не должна образовывать с используемым основанием - триэтиламином комплексного соединения, не способного к диссоциации. Данным требованиям максимально удовлетворяют галогениды цинка, которые использовались нами в качестве промоторов алкинилирования галогенидов германия.
Схема 1.1
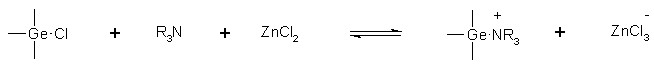
Нами найдено, что взаимодействие тетрахлорида германия с 1-алкинами в присутствии третичного амина и галогенида цинка приводит к образованию продуктов исчерпывающего алкинилирования – тетраалкинилгерманов 1a-d.
Схема 1.2
GeCl4 + 4 RCCH + 4 Et3N + 4 ZnX2 ![]() (RCC)4Ge + 4 Et3N+H•ZnCl3-
(RCC)4Ge + 4 Et3N+H•ZnCl3-
1a-d
R = Me3Si (1a), Ph (1b), CH2OAc (1c), n-Bu (1d)
Реакция легко протекает в среде эфирных растворителей – 1,4-диоксана или ТГФ. Использование этих сред обеспечивает доступность галогенида цинка, образующего растворимые эфираты.
При использовании низкокипящего триметилсилилацетилена реакцию целесообразно проводить в ТГФ или 1,4-диоксане при 50-55оС чтобы избежать потерь легколетучего реагента. Ход реакции хорошо контролируется тонкослойной или газожидкостной хроматографией по расходованию исходного 1-алкина.
В аналогичную реакцию удалось ввести и галогенид германия, содержащий органический радикал при атоме германия – фенилтрихлоргерман. Продуктом реакции явился фенил-трис(фенилэтинил)герман, который был получен с выходом 85% по схеме:
Схема 1.3
PhGeCl3 + 3 RCCH + 3 Et3N + 3 ZnX2 ![]() PhGe(CCR)3 + 3 Et3N+H•ZnCl3-
PhGe(CCR)3 + 3 Et3N+H•ZnCl3-
2a-c
R = n-Bu (2a), Me3Si (2b), Ph (2c)
Нами также обнаружено, что предлагаемый метод не может быть распространен на триалкилхлоргерманы и диалкилдихлоргерманы, которые не вступают в реакцию с 1-алкинами в присутствии эквимольных количеств галогенида цинка и третичного амина. Этот факт ставит под сомнение образование в условиях реакции в качестве интермедиатов алкинилидов цинка.
В тоже время, именно три- и тетрагалогениды германия, по-видимому, способны образовывать при взаимодействии с триэтиламином и хлоридом цинка достаточно реакционные электрофильные частицы. Присутствие двух или трех алкильных заместителей у атома германия резко снижает возможность образования активного германийсодержащего электрофила. Высокая электроотрицательность входящей алкинильной группы поддерживает сохранение электрофильных свойств атома германия и обеспечивает возможность исчерпывающего замещения атомов галогена в три- и тетрагалогенидах германия.
1.2 Взаимодействие тетра(диалкиламиноамино)германов и фенилтрис(диалкиламино)германов с 1-алкинами
Исследование поведение тетра(диалкиламино)германов при взаимодействии с 1-алкинами показало, что даже длительное нагревание тетра(диэтиламино)германа с фенилацетиленом при 150-180оС не приводит к образованию германийацетиленовых соединений, из реакционной массы можно практически количественно выделить перегонкой не вступивший во взаимодействие фенилацетилен. Успеха удается достичь при проведении реакции в присутствии эквимольного количества галогенида цинка, при этом с хорошими выходами образуются тетраалкинилгерманы.
Ход реакции легко контролируется с помощью ТСХ, по данным которой полная конверсия фенилацетилена наступает после 4 ч нагревания при 100оС.
Схема 1.4
Ge(NR2)4 + 4 RCCH + 4 ZnX2 ![]() (RCC)4Ge + 4 R2NH • ZnCl2
(RCC)4Ge + 4 R2NH • ZnCl2
3a-b 1a-g
R` = Me3Si (1a), Ph (1b), CH2OAc (1c), n-Bu (1d), n-Hex (1e), 4-BrC6H4 (1f), -Np (1g); X = Cl, Br;
В аналогичных условиях во взаимодействие с 1-алкинами в присутствии галогенида цинка вступает трис(диалкиламино)фенилгерман, образуя с хорошими выходами продукты алкинилирования:
Схема 1.5
PhGe(NR2)3 + 3 RCCH + 3 ZnX2 ![]() PhGe(CCR)3 + 3 R2NH•ZnCl2
PhGe(CCR)3 + 3 R2NH•ZnCl2
4a-b 2a-c
R` = n-Bu (2a), Me3Si (2b), Ph (2c); X = Cl, Br
Наиболее вероятно предположить, что гермилирующая система при использовании аминогерманов возникает благодаря координации галогенида цинка по атому азота аминогермана, что приводит к увеличению электрофильности атома германия и одновременно к формированию эффективной уходящей группы.
Схема 1.6

2 Синтез ацетиленовых производных кремния
2.1 Диалкиламиносиланы в процессах силилирования 1-алкинов
Для осуществления силилирования 1-алкинов необходима активация одного из участников данного процесса – кремнийорганического соединения или 1-алкина. И, если в процессе металлорганического синтеза, происходит активация нуклеофильных свойств 1-алкина за счет его депротонирования и превращения в реакционноспособный алкинил-анион, то в качестве альтернативы нам представлялось перспективным исследовать возможность активации гетероатомного кремнийорганического соединения по связи кремний-гетероатом, с целью усиления электрофильных свойств атома кремния. Наиболее подходящим объектом для этой цели нам представлялись аминосиланы, а в качестве способа активизации их электрофильных свойств – взаимодействие последних с кислотами Льюиса.
2.1.1 Синтез аминосиланов.
Для синтеза аминосиланов мы использовали взаимодействие первичных и вторичных аминов с органохлорсиланами, по схеме:
Схема 2.1
R1R2R3SiCl + 2 R`2NH ![]() R1R2R3SiNR`2 + R`2NH·HCl
R1R2R3SiNR`2 + R`2NH·HCl
5a-o
R1 = R2 = R3 = Me; R` = Me (5a), Et (5b), n-Bu (5c), n-Am (5d), пиперидил (5e), морфолил (5f); R1 = R2 = Me; R3 = Et, R` = пиперидил (5g); R1 = R2 = Me; R3 = n-Bu; R` = пиперидил (5h);
R1 = R2 = Me; R3 = Ph; R` = Et (5i); R1 = R2 = Me; R3 = n-BuO; R` = Et (5j);
R1 = R2 = Me; R3 = PhO; R` = Et (5k); R1 = R2 = Me; R3 = PhCC; R` = Et (5l), n-Bu (5m), пиперидил (5n), морфолил (5o).
R3SiCl + 2 R``NH2 ![]() R3SiNHR`` + R``NH2·HCl
R3SiNHR`` + R``NH2·HCl
5p-u
R = Me; R``= Et (5p), All (5q), t-Bu (5r), PhCH2 (5s), Ph (5t), PhNH (5u);
R1R2SiCl2 + 4 R`2NH ![]() R1R2Si(NR`2)2 + 2 R`2NH·HCl
R1R2Si(NR`2)2 + 2 R`2NH·HCl
6 a-d
R1 = R2 = Me; R` = Et (6a), n-Bu (6b), пиперидил (6c), морфолил (6d);
Во многих случаях препаративные методики синтеза этих соединений нами значительно улучшены за счет использования в реакции десяти-пятнадцатикратного избытка вторичного амина, заменяющего инертный растворитель.
Другим использованным в работе эффективным методом синтеза органоаминосиланов, была дегидроконденсация гидридсиланов с вторичными аминами, осуществлявшаяся в присутствии палладиевой черни, позволившая получать целевые аминосиланы с хорошими выходами из стерически затрудненных субстратов:
Схема 2.2
Pd
R1R2R3SiH + R`2NH ![]() R1R2R3SiNR`2 + H2
R1R2R3SiNR`2 + H2
5i, v-w
R1 = R2= Me, R3 = Ph; R` = Et (5i); R1 = R2 = Me, R3= 4-CF3-C6H5; R` = Et (5v);
R1 = R2= R3 = R` = Et (5w);
Удобным препаративным способом синтеза аминосиланов из аминов, обладающих высокой основностью и температурой кипения, является реакция переаминирования гексаметилдисилазана, использованная нами для наработки значительных количеств N-триметилсилилпиперидина.
2.1.2 Выбор оптимальных условий силилирования 1-алкинов аминосиланами.
Высокая реакционная способность аминосиланов по отношению к протонным кислотам, в том числе карбоновым кислотам, спиртам, тиолам, позволяет широко использовать их в качестве силилирующих реагентов, однако, диалкиламиносиланы оказались неспособными к взаимодействию с 1-алкинами даже при длительном нагревании до 200оС. Характер взаимодействия меняется при проведении реакции в присутствии галогенидов цинка. Начинающаяся при 25-400С и быстро протекающая при 80-1000С реакция приводит к селективному образованию силилацетиленов:
Схема 2.3
Me3SiNR`2 + PhCCН + ZnCl2 ![]() Me3SiCCPh + R2NH•ZnCl2
Me3SiCCPh + R2NH•ZnCl2
5a-f 7a
Далеко не все соединения, содержащие в своем составе связи Si-N, оказались способны вступать в аналогичную реакцию с ацетиленовыми соединениями. Так, полное отсутствие реакционной способности при взаимодействии с 1-алкинами, в присутствии галогенидов цинка, демонстрировали такие низкоосновные амины, как гексаметилдисилазан, N-силилированные амиды кислот, N-триметилсилиланилин, N-триметилсилилимидазол, N-триметилсилилфенилгидразин. Наиболее эффективными силилирующим агентами для 1-алкинов оказались триметилдиалкиламиносиланы, которые способны практически количественно силилировать 1-алкины в присутствии галогенидов цинка в 1,4-диоксане (Табл.1).
Таблица 1 - Силилирование фенилацетилена аминосиланами в присутствии ZnCl2 в 1,4-диоксане (соотношение реагирующих веществ 1:1.5:1.625, нагревание в течении 1.5 ч при 100оС)
| Аминосилан | Выход 7a, % | Аминосилан | Выход 7a, % |
| Me3SiNMe2 | 92 | Me2EtSiNEt2 | 82 |
| Me3SiNEt2 | 94 | Me3SiNHBu-t | 87 |
| Me3SiN(Bu-n)2 | 84 | Me3SiNHEt | 85 |
| Me3SiN(Am-n)2 | 80 | PhCH2NHSiMe3 | 54 |
| 94 | 86 | ||
| Et3SiNEt2 | 74 | Me2PhSiNEt2 | 90 |
| t-BuMe2SiNEt2 | 0 | Me2(n-Bu)SiNEt2 | 78 |
| Me3SiNHAll | 83 | (4-СF3-Ph)Me2SiNEt2 | 96 |
В качестве катализаторов нами был опробован обширный ряд кислот Льюиса, включивший BF3, BBr3, AlCl3, AlBr3, FeCl3, CoCl2, NiCl2, SbCl5, TiCl4, CuCl2, HgCl2, Однако, галогениды бора, алюминия, титана и олова вызывают быструю полимеризацию 1-алкинов или вступают в обменное взаимодействие с аминосиланом, а галогениды меди и ртути в присутствии аминосиланов быстро превращаются в алкинилиды соответствующих металлов, неспособные в данных условиях эффективно образовывать силилацетилены.
Представляется интересным тот факт, что только немногие кислоты Льюиса, в частности ZnCl2, ZnBr2, ZnI2, CdCl2, CdBr2, InBr3, и в меньшей степени Zn(OTf)2, способны эффективно содействовать силилированию 1-алкинов.
Таблица 2 – Эффективность кислот Льюиса в силилировании фенилацетилена N-триметилсилилдиэтиламином в среде 1,4-диоксана
| Кислота Льюиса | Выход 7a, % | Кислота Льюиса | Выход 7a, % |
| ZnBr2 | 99 | CdCl2 | 20 |
| ZnJ2 | 99 | CdBr2 | 28 |
| ZnF2 | 0 | CdJ2 | 40 |
| Zn(OTf)2 | 49 | InBr3 | 99 |
| Zn(OAc)2 | 0 | ZnSO4 | 0 |
Взаимодействие между диалкиламиносиланами и 1-алкинами может быть осуществлено как без растворителя, так и в инертных, по отношению к реагентам и образующимся в ходе реакции комплексам, растворителях – углеводородах, галогенуглеводородах, простых эфирах. Одним из наиболее удобных растворителей, обеспечивающих эффективное взаимодействие реагентов, является 1,4-диоксан, используемый при температуре его кипения. В тоже время, использование азотсодержащих донорных растворителей – амидов, нитрилов, алифатических и ароматических аминов неэффективно, что, по-видимому, объясняется их конкуренцией с аминосиланами в процессе донорно-акцепторного взаимодействия с галогенидами цинка, а также взаимодействием с активным кремнийорганическим электрофилом, образующимся из аминосилана и галогенида цинка (кадмия, индия).
Наилучшие выходы продукта силилирования получены при использовании не эквимольного количества аминосилана, а полуторократного избытка по отношения к теоретическому количеству и 25% избытка галогенида цинка по отношению к аминосилану.
Возможно предположить несколько вариантов участия галогенидов цинка в процессе прямого силилирования 1-алкинов, которые представлены на схеме 2.4.
Схема 2.4

Первые два из них (путь А и В, соответственно) предполагают первоначальное взаимодействие галогенидов цинка, выступающих в данном случае в качестве кислоты Льюиса, с -электронами тройной связи. Причем, в соответствии со схемой А, образующийся при взаимодействии 1-алкина с галогенидом цинка комплекс -V типа (I), под действием аминосилана подвергается депротонированию с последующим образованием галогенида алкинилцинка (II). Ключевой стадией пути А является взаимодействие алкинилцинка с диалкиламиносиланом с образованием продукта силилирования (III). Однако, полученные нами экспериментальные данные противоречат нуклеофильному механизму силилирования Данная схема предполагает в качестве стадии, определяющей скорость процесса, стадию депротонирования 1-алкина. Однако, проведенный нами кинетический эксперимент, в ходе которого сравнивалась реакционная способность замещенных фенилацетиленов, имеющих как электронодонорные, так и электроноакцепторные заместители (см. табл. 4), однозначно демонстрирует большую реакционную способность субстратов, имеющих электронодонорные заместители, что противоречит их способности к депротонированию.
Таблица 4 - Влияние кислотности 1-алкина на эффективность силилирования арилацетиленов под действием N-триметилсилилдиэтиламина и ZnCl2 (1,4-диоксан, 100оС, 1 час)
| 1-Алкин | Выход 4-R-C6H4CC-SiMe3 Выход 4-H-C6H4CC-SiMe3 |
| 4-Me-C6H4CCH | 1,52 |
| 4-MeO-C6H4CCH | 1,48 |
| 4-H-C6H4CCH | 1,00 |
| 4-CF3-C6H4CCH | 0,49 |
| 4-NO2-C6H4CCH | 0,38 |
Специально проведенные контрольные опыты показали отсутствие взаимодействия между полученными независимым методом PhCCZnCl и диалкиламиносиланами.
Практически те же соображения противоречат и возможности реализации пути В, в соответствии с которой, первоначально образующийся при взаимодействии 1-алкина и галогенида цинка -комплекс (IV), превращается в -связанный цвиттер-ион (V), подвергающийся далее переметаллированию под действием диалкиламиносилана.
Экспериментальные данные хорошо согласуются со схемой, предполагающей в качестве ключевой стадии взаимодействия электрофильную атаку кремнийсодержащего комплекса по тройной связи 1-алкина (путь С). В соответствии с ней, галогенид цинка вступает во взаимодействие с электронной парой диалкиламиносилана, с образованием комплекса n-V типа, в котором электрофильные свойства атома кремния резко возрастают и он получает способность к прямому взаимодействию с тройной связью (интермедиат VI). Результатом взаимодействия является силилсодержащий винилкатион (V), стабилизирующийся в результате выброса протона, с образованием продукта реакции. Данная схема полностью согласуется как с результатами сравнительного кинетического исследования реакционной способности 1-алкинов, так и с данными по реакционной способности различных аминосиланов (табл.1).
2.1.3 Синтез триорганоалкинилсиланов.
В качестве субстратов силилирования нами исследовались различные соединения, содержащие в своем составе терминальную ацетиленовую связь.
Изучение силилирования самого ацетилена проводили барботируя очищенный ацетилен через раствор диэтиламинотриметилсилана и ZnCl2 (мольное соотношение 1:1.5, концентрация по аминосилану 0,5 моль/л) в ТГФ при 50оС в течении 10 часов. ГХ/МС анализ реакционной массы показал наличие двух продуктов – триметилсилилацетилена 7b и бис-(триметилсилил)ацетилена 7c, которые удалось выделить из реакционной массы после гидролиза и перегонки с выходом 22% и 8% соответственно.
Схема 2.5

7b 7c
По-видимому, столь низкий выход целевого продукта объясняется ничтожно малой растворимостью газообразного ацетилена в реакционной массе, его высокой кислотностью, а также возможным «выносом» легколетучего продукта сильным током ацетилена.
Гораздо лучших результатов удалось достичь при силилировании гомологов ацетилена, а также различных арилацетиленов. При этом все эксперименты проводили при использовании избытка силилирущего реагента – полуторократного мольного количества аминосилана и 25% избытка галогенида цинка по отношению к аминосилану. Ход реакции в большинстве случаев легко контролируется ТСХ по расходу исходного ацетилена. Для достижения максимального выхода силилалкина требуется нагревание при 100оС в течении 1,5-3 часов. В случае использования легколетучих триметилсилилацетилена и трет-бутилацетилена время реакции увеличивается до 6-8 часов в связи с невозможностью проведения реакции в указанном температурном режиме из-за возможной потери исходного 1-алкина.
Схема 2.6

7d-s
R = n-Bu (7d), n-Am (7e), n-Hex (7f), n-Oc (7h), t-Bu (7i), Me3Si (7j), 4-Me-Ph (7k), 4-CF3-Ph (7l), 4-MeOCO-Ph (7m), 4-MeO (7n), 4-CN-Ph (7o), 4-NO2-Ph (7p), 4-Cl-Ph (7q), 4-Br-Ph (7r), -Np (7s)
На примере октина-1 показана возможность осуществления реакции без использования растворителя, однако такой вариант не очень удобен из-за невозможности эффективно перемешивать реакционную массу, что может негативно сказываться на величине выхода целевого продукта.
Силилирование 1-алкинов, содержащих пропаргильный фрагмент в большинстве случаев протекает с хорошими выходами. Таким образом, было проведено силилирование целого ряда простых и сложных эфиров пропаргилового спирта, в том числе содержащих различные функциональные группы.
Схема 2.7

8a-p
X = OAc (8a), OCOPh (8b), OCOPh-4-Me (8c), OCOPh-3,5-(NO2)2 (8d), OCOCH=CHPh (8e), OCOCH2Cl (8f), OCOCHCl2 (8g),OCOCCl3 (8h), OCOBu-i (8i), OCOCH=CHCH3 (8j), Cl (8k), Br (8l), OPh (8m), OPh-2-NO2 (8n), OPh-4-NO2 (8o), OCH2Ph (8p)
К сожалению, не удалось достигнуть успеха при силилировании ацетиленовых альдегидов, кетонов и их производных. Взаимодействие пропиолового альдегида и метилэтинилкетона с силилирующей системой даже в мягких условиях сопровождается сильным осмолением реакционной массы. Аналогично ведут себя и продукты защиты карбонильной группы – диэтилацеталь пропиолового альдегида и 2-метил-2-этинил-1,3-диоксан.
Особый интерес представляет силилирование ацетиленовых спиртов. Силилирование пропаргилового спирта может быть осуществлено напрямую, при использовании большого избытка силилирующей системы, однако выход в этом случае весьма умеренный и реакция сопровождается заметным осмолением:
Схема 2.8
1. 5e 3 моль, ZnCl2 3.6 моль
1,4-диоксан, 100оС, 4 ч
HCCCH2OH ![]() Me3SiCCCH2OH
Me3SiCCCH2OH
2. HCl 2 моль/л 32 %
Предварительная защита гидроксильной группы с использованием стандартных приемов обеспечивает гладкое протекание реакции при использовании ацетатной и бензильной защиты. Наилучшие выходы получены при использовании триметилсилильной защиты, которая является самой приемлемой в плане легкости введения и последующего удаления:
Схема 2.9

9a-d
R = R` = H (9a); R = Me, R` = H (9b); R = R` = Me (9c); R = Ph, R` = H (9d)
Гидролиз реакционной массы в мягких условиях – охлажденным раствором NH4Cl позволяет выделить 9a-d.
Использование таких распространенных защитных групп как тетрагидропиранильная и винилэтильная не увенчалось успехом. Проведение силилирования 2-(пропин-2-ил-1-окси)тетрагидропирана и 3-(1-этоксиэтокси)пропина-1 сопровождалось сильным осмолением реакционной массы.
Силилирование пропиоловой кислоты, как и силилирование пропаргилового спирта возможно при использовании трех эквивалентов силилирующей системы, однако низкий выход (24 %) целевого продукта обуславливает необходимость предварительной защиты карбоксильной группы посредством получения триметилсилилового или этилового эфира:
Схема 2.10

10a-c
R = H (10a), SiMe3 (10b), Et (10c)
Как и следовало ожидать, в связи с электрофильным характером силилирования, наиболее толерантными субстратами оказались ацетиленовые углеводороды, в особенности алкилацетилены, фенилацетилен и его замещенные, сложные эфиры пропаргилового спирта и карбоновых кислот. Гораздо менее реакционными оказались ацетиленовые субстраты, содержащие при тройной связи электроноакцепторную группировку, в том числе сложные эфиры пропиоловой кислоты, хлористый и бромистый пропаргил.
2.1.4 Синтез диалкинилсиланов
Взаимодействие диорганоди(диалкиламино)силанов 6a-e с 1-алкинами осуществляли в условиях, аналогичных используемым в реакции триорганоаминосиланов, однако соотношение реагентов в данном случае иное. Если при получении алкинилтриорганосиланов использовался полуторократный избыток силилирющего реагента, то в данном случае соотношение было эквимольным. Это обусловлено тем, что образующиеся после гидролиза линейные и циклические силоксаны могут затруднить выделение целевого продукта. Наилучшие выходы – до 74% - целевых диалкинилсиланов получены при проведении реакции в среде 1,4-диоксана в течении 3-4 часов.
Схема 2.11
R1R2Si(NR`2)2 + RCCH + 2 ZnCl2![]() R1R2Si(CCR)2 + 2 R2NH·ZnCl2
R1R2Si(CCR)2 + 2 R2NH·ZnCl2
6a-e 11a-f
R1 = R2 = Me; R` = Et, n-Bu, пиперидил, морфолил; R = Ph (11a);
R1 = R2 = Me; R` = пиперидил; R = 4-Br-Ph (11b), R = n-Bu (11c), Me3Si (11d), CH2OCOCH2Cl (11e); R1 = Ph, R2 = Me; R` = Et; R = Ph (11f);
Вещества 11а,с выделены после традиционной обработки реакционной массы вакуумной перегонкой, вещества 11b,d-f колоночной хроматографией на силикагеле с использованием хлороформа в качестве элюента или перекристаллизацией из гексана.
Ряд несимметричных диалкинилсиланов удалось синтезировать силилированием 1-алкинов диметил(фенилэтинил)аминосиланами 5l-o. В данном случае также пришлось использовать эквимольное соотношение реагентов, т.к. избыточное количество аминосилана после гидролиза превратилось бы в тетраметилдифенилэтинилдисилоксан, который затруднил бы выделение в чистом виде целевых продуктов.
2.2 Силилирование 1-алкинов галогенсиланами
Предпринятые нами попытки активирования галогенсиланов теми же приемами, что использовались для амино-, галогенгерманов и аминосиланов также оказались успешными и в этом случае. Взаимодействие триметилхлорсилана с фенилацетиленом, в присутствии хлорида цинка и триэтиламина, осуществленное в 1,4-диоксане, привело к образованию продукта силилирования с выходом 62%. Замена атома галогена в галогениде цинка и галогенсилане на бром или иод способствует существенному увеличению выхода целевого продукта – так, например, использование комбинации триметилбром(иод)силан : иодид цинка обеспечивает практически количественное протекание реакции, при этом время, необходимое для достижения максимального выхода также существенно снижается.
Схема 2.12
R1R2R3SiX + RCCH + ZnХ2 + Et3N ![]() R1R2R3SiCCR + Et3NH+ZnХ-3
R1R2R3SiCCR + Et3NH+ZnХ-3
7
R1 = R2 = R3 = Me; Х = Cl, Br, I; R = Ph (7a), n-Bu (7d), Me3Si (7j)
R1 = Ph, R2 = R3 = Me, Х = Cl, R = Ph (7t)
Кроме галогенидов цинка способность эффективно промотировать взаимодействие галогенсиланов с 1-алкинами показал InBr3. Применение других кислот Льюиса в данной реакции не эффективно. Реакция гладко протекает в среде 1,4-диоксана, несколько хуже при использовании ароматических или галогенированных углеводородов.
При эквивалентном соотношении реагентов в реакцию вступают диорганодихлор- и органотрихлорсиланы, при этом удается с хорошими выходами получать соответствующие диалкинилсиланы 11 (выход до 65%) и триалкинилсиланы 12 (выход до 56%):
Схема 2.13
RnSiCl4-n + n R`CCH + n ZnХ2 + n Et3N ![]() RnSi(CCR`)4-n + n Et3NH+ZnХ-3
RnSi(CCR`)4-n + n Et3NH+ZnХ-3
11a-d, 12a,b
n = 2, R = Me, R` = Ph (11a), 4-Br-C6H4 (11b), n-Bu (11c), Me3Si (11d);
n = 1, R = Me (12a), Et (12b); R` = Ph
Повысить выход ди- и триалкинилсиланов путем использования диорганодииод- и органотрииодсиланов не представлялось возможны из-за относительной труднодоступности и неустойчивости последних.
2.3 Силилирование 1-алкинов О-силилуретанами.
Попытка проведения силилирования фенилацетилена под действием О-триметилсилилпиперидинкарбоксилата 13 в условиях, аналогичных силилированию аминосиланами, не привела к получению 1-триметилсилилфенилацетилена даже в следовых количествах. Можно предположить, что комплекс, образующийся при взаимодействии ZnCl2 с 1,4-диоксаном, не способен к дальнейшему эффективному взаимодействию с O-силилкарбаматом. Лучших результатов удалось достичь при проведении реакции в среде углеводородных или хлорированных растворителей. Так, при использовании толуола, взаимодействие силилкарбамата с фенилацетиленом приводит к образованию триметилсилилфенилацетилена 7а с выходом 32%, а при использовании 1,2-дихлорэтана - 40%.
Схема 2.14
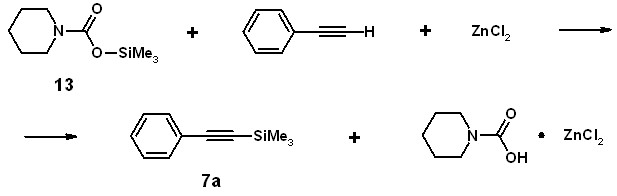
При этом выход целевого продукта увеличивается при использовании двукратного количества галогенида цинка.
Замена галогенида цинка на трибромид индия, привела к получению силилалкина с выходом 73% даже при эквимольном соотношении реагентов. Использование двукратного количества трибромида индия приводит к получению целевого продукта с практически количественным выходом.
Таблица 5 - Влияние условий реакции на взаимодействие фенилацетилена с О-триметилсилилпиперидинкарбоксилатом*
| Растворитель | Кислота Льюиса | Выход PhCCSiMe3, % |
| 1,4-диоксан | ZnCl2, 1 экв. | 0 |
| 1,4-диоксан | ZnCl2, 2 экв. | 0 |
| толуол | ZnI2, 1 экв. | 32 |
| толуол | ZnI2, 2 экв. | 48 |
| 1,2-дихлорэтан | ZnCl2, 1 экв. | 40 |
| 1,2-дихлорэтан | ZnCl2, 2 экв. | 52 |
| 1,2-дихлорэтан | InBr3, 1 экв. | 73 |
| 1,2-дихлорэтан | InBr3, 2 экв. | 98 |
* - Эквимольное соотношение реагентов, время реакции 3 ч при температуре кипения растворителя
Описанное превращение представляется весьма интересным, как в теоретическом, так и в практическом плане. Во-первых, это один из немногих примеров силилирования 1-алкинов под действием соединений кремния, содержащих связь Si-O. Во-вторых, сам факт эффективного силилирования 1-алкинов под действием О-силилкарбаматов при содействии InBr3 позволяет надеяться на то, что данный реагент может быть использован и в других реакциях силилирования субстратов, чувствительных к электрофильной атаке.
Нам представляется наиболее вероятным, что химизм данного процесса заключается в образовании электрофильной частицы (IIIIIIV) с электронодефицитным атомом кремния, как это представлено на схеме 2.15. Следующая за этим атака образовавшегося электрофила по тройной связи 1-алкина, приводит к винилкарбокатиону V, который далее стабилизируется путем выброса протона и образования силилалкина.
Схема 2.15

2.4 Использование тетрафенилэтинилаланата лития в синтезе алкинилсиланов.
Наряду с исследованием процессов прямого алкинилирования кремнийорганических соединений под действием 1-алкинов мы проводили поиск эффективных систем, позволяющих проводить металлирование терминальных алкинов, с последующим вовлечением образующихся металлорганических соединений в реакции с гетероатомными кремнийорганических соединений для получения силилалкинов.
Нам представлялось интересным изучить возможность использования тетраалкинилаланатов лития в реакциях алкинилирования кремнийорганических субстратов.
Взаимодействие алюмогидрида лития с фенилацетиленом протекает количественно при нагревании реагентов с среде ТГФ при температуре кипения в течении 30 мин:
Схема 2.16
LiAlH4 + 4 PhCCH ![]() LiAl(CCPh)4 + 2 H2
LiAl(CCPh)4 + 2 H2
В числе кремнийорганических субстратов нами был исследован ряд веществ, охватывающий наиболее доступные классы кремнийорганических соединений: галогенсиланы – триметилхлорсилан и триметилбромсилан, соединения, содержащие связь Si-N - гексаметилдисилазан, N-триметилсилилпиперидин и N-триметилсилилимидазол, соединения, содержащих связь Si-O - гексаметилдисилоксан, триметилсилилацетат, триметилсилилбензоат, N,O-бис(триметилсилил)ацетамид, пиперидинкарбамоилокситриметилсилан.
Исследование алкинилирования кремнийорганических субстратов проводили по общей схеме: в атмосфере аргона к раствору LiAl(CCPh)4 (1 экв.) в ТГФ прибавляли кремнийорганическое соединение (0,25 экв.), нагревали реакционную массу при перемешивании и температуре кипения растворителя. За ходом реакции наблюдали, отбирая пробы реакционной массы, которые анализировали на хромато-масс-спектрометре после предварительного гидролиза. Во всех случаях в реакционной массе присутствовали только фенилацетилен и 1-триметилсилилфенилацетилен, продукты восстановления отсутствовали. Препаративно продукт выделяли перегонкой в вакууме после обработки реакционной массы 5% HCl.
Алкинилирование галогенсодержащих субстратов протекало при нагревании в течении 5 часов, выход продукта алкинилирования увеличивался при замене атома хлора на бром, однако при использовании Me3SiBr процесс осложнялся побочной реакцией расщепления эфирной связи в ТГФ. Использование в качестве растворителя устойчивого к расщеплению 1,4-диоксана позволило увеличить выход целевого продукта до 80%.
Таблица 6 - Алкинилирование кремнийорганических субстратов тетрафенилэтинилаланатом лития в ТГФ
| Кремнийорганический субстрат | Выход 7a, % | Кремнийорганический субстрат | Выход 7a, % |
| Me3SiСl | 32 | Me3SiBr | 47 |
| 73 |  | 54 | |
| 80 | 95 |
Изучение поведения азотсодержащих кремнийорганических соединений показало их инертность в исследуемой реакции, после разложения реакционной массы раствором соляной кислоты в реакционной смеси присутствовали только исходный фенилацетилен и продукты гидролиза - гексаметилдисилоксан и соответствующий амин.
Наиболее активными оказались силилпроизводные различных кислот, содержащие связь Si-O, прибавление кремнийорганического соединения в этом случае сопровождалось сильным экзотермическим эффектом, а максимальный выход целевого вещества достигался через 1,5-2 часа нагревания при температуре кипения. Наилучшую активность показал О-силилуретан, выход целевого продукта в этом случае составил более 90%.
Гексаметилдисилоксан оказался нереакционноспособным в этих условиях.
Полученные результаты свидетельствуют о высокой реакционной способности тетрафенилэтинилалюмината лития по отношению к некоторым кремнийорганическим соединениям. Дальнейшие детальные исследования химических превращений тетраалкинилаланатов в реакциях с органическими и элементоорганическими субстратами могут оказаться весьма продуктивными в плане развития методов органического синтеза.
Структура всех полученных соединений подтверждена методами ЯМР 1Н, 13С, ИК-спектроскопии и масс-спектрометри, а также путем сравнения физико-химических констант с известными из литературы.
ВЫВОДЫ
1. Изучено взаимодействие 1-алкинов с галоген- и азотсодержащими силанами и германами в условиях активации реакций кислотами Льюиса. Установлена высокая реакционная способность тетрахлорида германия, органотрихлоргерманов, органогалогенсиланов, диалкиламиноорганосиланов и О-силилуретанов в присутствии галогенидов цинка или трибромида индия при взаимодействии с 1-алкинами с образованием соответствующих алкинилгерманов и алкинилсиланов.
2. Исследованы реакции 1-алкинов с тетрахлоридом германия, тетракис(диалкиламино)германами, органилтригалогенидами германия и органилтрис(диалкиламино)германами в присутствии галогенидов цинка. Показана возможность использования данных реакций для препаративного синтеза три- и тетраалкинилидов германия.
3. Выявлена способность различных моно- и диаминосиланов к эффективному взаимодействию с 1-алкинами в присутствии галогенидов цинка, кадмия или индия, приводящему к образованию силилалкинов. Предложен химизм реакции, включающий стадию электрофильного силилирования тройной связи. Разработана препаративная методика силилирования 1-алкинов.
4. Изучено взаимодействие органогалогенсиланов с 1-алкинами в присутствии галогенидов цинка или индия и триэтиламина. Определены условия, при которых наблюдается максимальная степень конверсии 1-алкинов в соответствующие силилпроизводные.
5. Выявлены и описаны различия в реакционной способности органических гетероатомных соединений кремния со связями Si-Hal, Si-N и Si-O при взаимодействии с тетраалкинилаланатами лития.
6. Разработаны оригинальные препаративные методики синтеза широкого ряда кремний- и германийацетиленовых соединений – алкинилтриорганосиланов, диалкинилдиорганосиланов, алкилтриалкинилсиланов, три- и тетраалкинилгерманов, в том числе, содержащих различные функциональные группы.
Список основных работ, опубликованных по теме диссертации
1. Andreev А.А., Konshin V.V., Komarov N.V., Rubin M., Brouwer C., Gevorgyan V. Direct electrophilic silylation of terminal alkynes // Organic Letters.- Vol.6.- №3.- 2004.- Р. 421-424.
2. Андреев А.А., Коншин В.В., Винокуров Н. А. Комаров Н.В. Синтез три- и тетраалкинилгерманов // Известия Академии наук. Серия химическая.- 2006.- №8.- С. 1377-1379.
3. Коншин В.В., Андреев А.А. Сила-де-гидрирование 1-алкинов // Сб. науч. тр. «Успехи в химии и химической технологии».- М.: РХТУ им Д.И.Менделеева.- 2004.- т.18.- № 8 (48).- С.30-32.
4. Андреев А.А., Коншин В.В., Комаров Н.В. Новые реакции, приводящие к образованию кремний-углеродной связи // Наука Кубани.- 2007.- №2.- С. 10-13.
5. Андреев А.А., Коншин В.В., Комаров Н.В. Новые подходы к синтезу ацетиленовых производных элементов. IVb группы // XVII Менделеевский съезда по общей и прикладной химии: тез. докл. 21-26 сентября 2003.- Казань, 2003.- С.85.
6. Коншин В.В., Ольховская Л.И., Шиликова Е.В., Винокуров Н. А., Андреев А.А., Комаров Н.В. Аминосиланы в реакциях силилирования 1-алкинов // VII Школа-конференция по органической химии: тез. докл. Екатеринбург.-2004.- С. 262.
7. Коншин В.В., Андреев А.А., Комаров Н.В. Аминосиланы в реакциях силилирования простых и сложных пропаргиловых эфиров// VII Школа-конференция по органической химии: тез. докл. Екатеринбург.-2004.- С. 36.
8. Андреев А.А., Коншин В.В., Комаров Н.В. Новые пути синтеза крамнийацетиленовых соединений // 10 Всероссийская конференция «Кремнийорганические соединения: синтез, свойства, применение»: тез. докл. 26-30 мая 2005 г.- Москва.- С. У-25.
9. Коншин В.В., Андреев А.А., Комаров Н.В. Взаимодействие галогенсиланов с 1- алкинами // VIII Молодежная научная школа-конференция по органической химии: тез. докл. 22-26 июня 2005 г.- Казань.- С.64.
10. Коншин В.В., Андреев А.А., Комаров Н.В. Взаимодействие триметилгалогенсиланов с 1-алкинами. // ХV Российская молодежная конференция «Проблемы теоретической и экспериментальной химии»: тез. докл. 19-22 апреля 2005, г. Екатеринбург.- С. 308.
11. Коншин В.В., Ольховская Л.И., Андреев А.А.Комаров Н.В. Синтез бисалкинилорганосиланов // ХVI Российская молодежная научная конференция «Проблемы теоретической и экспериментальной химии»: тез. докл. 25-28 апреля 2006, г.Екатеринбург.- С. 315-316.
12. Коншин В.В., Андреев А.А., Комаров Н.В. Синтез три- и тетраалкинилгерманов // ХVI Российская молодежная научная конференция «Проблемы теоретической и экспериментальной химии»: тез. докл. 25-28 апреля 2006, г.Екатеринбург.- С. 314-315.
13. Коншин В.В., Андреев А.А., Комаров Н.В. Реакция тетрахлорида германия и органилтрихлоргерманов с 1-алкинами // Материалы IV Международной научной конференции «Химия, химическая технология и биотехнология на рубеже тысячелетий».- Томск: изд. ТПУ.- 2006.- С. 358.
14. Коншин В.В., Андреев А.А., Комаров Н.В. Взаимодействие О-силилуретанов с 1-алкинами // IX Научная школа–конференция по органической химии: тез. докл. 11-15 декабря 2006, г. Москва.- 2006.- С. 201.
15. Андреев А.А., Коншин В.В., Комаров Н.В. Новые подходы к синтезу ацетиленовых производных кремния и германия // Конференция грантодержателей регионального конкурса РФФИ и администрации Краснодарского края «Юг России» «Вклад фундаментальных исследований в развитие современной инновационной экономики Краснодарского края»: тез. докл.- Краснодар.- 2006.- С. 42.
16 Андреев А.А., Коншин В.В., Рыжкова Н.А., Комаров Н.В. Новые реакции приводящие к образованию силил- и гермилацетиленов // XVIII Менделеевский съезд по общей и прикладной химии: тез. докл. 23-28 сентября 2007, г. Москва.- С. 99.
17. Коншин В.В., Андреев А.А., Комаров Н.В. Силилирование ацетиленовых спиртов // XVII Российская молодежная научная конференция «Проблемы теоретической и экспериментальной химии»: тез. докл. 17-20 апреля 2007, г. Екатеринбург.- С. 288.
18. Патент РФ № 2368615. Способ получения алкинилсиланов. / А.А. Андреев, В.В. Коншин, Н.В. Комаров; заявитель и патентообладатель ГОУ ВПО КубГУ.- №2008131242/04; заявл. 28. 07. 2008; опубл. 27.09.2009, Бюл. № 27.
Автореферат
диссертации на соискание ученой степени
кандидата химических наук
Специальность: 02.00.03 – Органическая химия
Коншин Валерий Викторович
______________________________________________
Центр «Универсервис»
Кубанский Государственный университет
350040, г. Краснодар, ул. Ставропольская, 149
Подписано в печать 18 ноября 2009, формат 60x84 1/16
Заказ№699, Тираж 100
