

спектроскопия высокого разрешения и внутренняя динамика молекул
На правах рукописи
Бехтерева Елена Сергеевна
СПЕКТРОСКОПИЯ ВЫСОКОГО РАЗРЕШЕНИЯ И ВНУТРЕННЯЯ
ДИНАМИКА МОЛЕКУЛ
01.04.02 — теоретическая физика
Автореферат
диссертации на соискание ученой степени
доктора физико-математических наук
Томск 2008
Работа выполнена на кафедре оптики и спектроскопии ГОУ ВПО «Томский государственный университет»
Научный консультант: доктор физико-математических наук, профессор
Улеников Олег Николаевич
Официальные оппоненты: доктор физико-математических наук,
Быков Александр Дмитриевич
доктор физико-математических наук, профессор
Килин Виктор Андреевич
доктор физико-математических наук, профессор
Самсонов Борис Федорович
Ведущая организация: Институт прикладной физики РАН,
г. Нижний Новгород
Защита состоится «22» мая 2008 г. в 14 часов 30 мин. на заседании диссертационного совета Д 212.267.07 при Томском государственном университете по адресу: 634050, г. Томск, пр. Ленина 36, аудитория 119.
С диссертацией можно ознакомиться в Научной библиотеке Томского государственного университета по адресу: г. Томск, пр. Ленина, 34а.
Автореферат разослан «xx» xxxxx 2008 г.
Ученый секретарь
диссертационного совета,
д.ф.-м.н. И.В. Ивонин
общая характеристика работы
Актуальность темы. Высокий уровень развития современной теоретической колебательно-вращательной спектроскопии молекул позволяет адекватно интерпретировать полученную экспериментальную информацию и путем изучения тонкой структуры спектров определять такие характеристики молекул, которые открывают возможность для исследования более сложных эффектов внутримолекулярной природы.
Колебательно-вращательные спектры высокого разрешения являются наиболее полным и надежным источником информации о характере внутримолекулярных взаимодействий, состояниях и свойствах молекул. Определяемые из эксперимента параметры спектральных линий содержат информацию о таких важнейших характеристиках молекул, как структурные постоянные, внутримолекулярное силовое поле, межмолекулярный потенциал, электрический и магнитный моменты.
Известно, что внутренняя динамика молекулы в основном определяется внутримолекулярной потенциальной функцией. Поэтому важным условием понимания протекающих в молекуле процессов является получение корректной информации о последней. В связи со сказанным становится понятен интерес к количественному определению параметров потенциальных функций молекул.
Методы решения отмеченной проблемы развиваются в физике и химии в течение уже многих лет. В этой связи следует отметить две основных тенденции в решении данной проблемы. С одной стороны, в течение уже нескольких десятков лет ведутся интенсивные попытки решения данной проблемы с помощью ab initio методов. Однако вплоть до настоящего времени точность таких расчетов для молекул с числом атомов более трех всё ещё остаётся на четыре - пять порядков хуже экспериментальных точностей в определении тех наблюдаемых величин, которые могут быть получены на основе информации о внутримолекулярной потенциальной функции. Точность расчета может быть, в принципе, улучшена, но это требует настолько резкого увеличения времени счёта даже на современных суперкомпьютерах, а, следовательно, резкого увеличения финансовых затрат, что задача становится практически нереализуемой в настоящее время,
Альтернативный подход к решению задачи определения многомерных потенциальных поверхностей молекул может быть назван полуэмпирическим. Он тоже развивается в течение уже многих десятков лет и основан на использовании, с одной стороны, прецизионной экспериментальной информации о молекулярных спектрах высокого разрешения инфракрасного, видимого и микроволнового диапазонов, а, с другой стороны, результатов колебательно - вращательной теории многоатомных молекул. В отличие от ab initio методов, полуэмпирические методы позволяют получать информацию о параметрах потенциальной поверхности с весьма высокой точностью. Как следствие, в последние годы в ряде научных центров, занимающихся исследованием физики молекул (Университет Вупперталя, Вупперталь, Германия; Университет наук и технологий, Хэфей, Китай; Высшая технологическая школа, Лондон, Великобритания; ЕТН, Цюрих, Швейцария; Университет Бургундии, Дижон, Франция и др.), предпринимались попытки развить новые высокоточные методы количественного описания многомерных энергетических поверхностей молекул. Среди полуэмпирических можно назвать вариационные методы, в частности MORBID-метод, разработанный Йенсеном, расчёты, основанные на методе Монте-Карло и расчёты, использующие преобразование Ван-Флека. Не обсуждая здесь их детально, отметим лишь, что все они наряду с достоинствами обладают и теми или иными недостатками. В частности, вариационные методы, позволяя получить высокоточные результаты, реально применимы только к самым малым трёх- (в ряде случаев четырёх- ) атомным молекулам и, более того, требуют для своей реализации достаточного большого времени компьютерного счёта и компьютерных ресурсов. Упомянутые недостатки легко объяснимы, если вспомнить что любой вариационный метод – это итерационная процедура, нуждающаяся в прямом многократном построении матрицы колебательно - вращательного гамильтониана в том или ином базисе и её диагонализации. Как показывает опыт спектроскопии, для достижения достаточных точностей в определении параметров внутримолекулярной потенциальной функции необходимо учесть до 10-15 квантов возбуждения на каждую колебательную степень свободы. Попытки учета также и вращательных степеней свободы приводят к многократному росту размеров используемых матриц. В итоге получаются матрицы размерами до 107 – 108 для четырёх-пяти атомных молекул. Как следствие, можно сказать, что используемые в настоящее время в вариационных методах модели гамильтониана и расчетные схемы применимы лишь к самым малым молекулам. Аналогичные плюсы и минусы можно отметить и для рассчётов основанных на методе Монте-Карло.
В связи с вышесказанным, в течение последних лет в лаборатории молекулярной спектроскопии Томского госуниверситета разрабатывались различные подходы к реализации обратных спектроскопических задач и методы определения внутримолекулярной потенциальной функции многоатомных молекул, которые позволили бы совместить в себе преимущества упомянутых выше подходов и, вместе с тем, были бы в значительной степени свободны от их недостатков. Следует также отметить независимость разрабатываемых методов от типа симметрии молекул и количества ядер в ней. Другими словами, они, практически без изменений, могут быть применены не только к трёх - пятиатомным молекулам, но и к молекулам с большим числом атомов.
Говоря о полуэмпирических методах, следует отметить, что задачу определения потенциальной функции разумно разбить на две большие подзадачи:
- разработка методов и непосредственное определение на их основе параметров внутримолекулярной потенциальной функции конкретных молекул;
- анализ спектров высокого разрешения для обеспечения первой подзадачи исходной высокоточной экспериментальной информацией.
В связи с этим в данной работе можно выделить три основных комплекса решаемых задач. Это:
- разработка эффективных, простых и, вместе с тем, применимых не только к малым, но и к молекулам с произвольным числом атомов и различной симметрией методов определения внутримолекулярной потенциальной функции;
- на этой основе разработка алгоритмов и создание программ реализации развитых
методов определения ВМПФ для различного типа многоатомных молекул;
- анализ спектров высокого разрешения для обеспечения первой подзадачи исходной высокоточной экспериментальной информацией.
Следует заметить, что для реализации последнего комплекса проблем возникает необходимость модификации известных и разработки новых методов как интерпретации колебательно-вращательных спектров, так и методов решения обратных спектроскопических задач различного типа многоатомных молекул.
Перечисленная выше совокупность проблем современной теоретической спектроскопии, их практическая значимость для многочисленных задач астрофизики, атмосферной оптики, физики и химии молекул и др. – все это в совокупности определяет актуальность исследований выполненных в данной диссертационной работе.
В свете вышесказанного основными целями данной работы являлись:
- разработка эффективного, простого и, вместе с тем, применимым не только к малым, но и к молекулам с произвольным числом атомов и различной симметрией методов определения внутримолекулярной потенциальной функции;
- на этой основе разработка алгоритмов и создание программ реализации развитых
методов определения ВМПФ для различного типа многоатомных молекул;
- анализ спектров высокого разрешения для обеспечения первой подзадачи исходной высокоточной экспериментальной информацией. Реализация последнего предполагает разработку новых и усовершенствование используемых в настоящее время в теоретической молекулярной спектроскопии методов и моделей, которые бы позволили корректно описывать сложную структуру современных колебательно – вращательных спектров высокого разрешения в условиях многочисленных и сильных резонансных взаимодействий, неоднозначностей и сильных корреляций между параметрами моделей.
Конкретная реализация указанных целей исследования включала в себя решение следующих задач:
- На основе исследования трансформационных свойств различных колебательных координат, использующихся в колебательно – вращательной спектроскопии многоатомных молекул, построение точного оператора для «кинетической» части гамильтониана молекулы, который, не уступая в корректности так называемым ЕКЕ («exact kinetic energy») операторам, (а). был бы применим не только к молекулам с небольшим числом атомов, но к произвольным многоатомным молекулам; (б). был бы проще ЕКЕ-операторов при практических расчетах; (в). в рамках унифицированного подхода был бы применим к любым изотопическим модификациям рассматриваемой молекулы.
- На основе использования методов операторной теории возмущений и полученного гамильтониана многоатомной молекулы разработка эффективного алгоритма, который позволил бы избежать построения и последующей диагонализации матриц большой размерности. Данное условие является основополагающим для возможности применения результатов данной работы не только к малым, но и к любым многоатомным нормальным молекулам.
- На основе результатов решения вышеназванных задач, разработка простого и, вместе с тем, эффективного метода определения ВМПФ нормальных молекул и его апробация на трех-, четырех- и пяти-атомных молекулах различной симметрии.
- Модернизация и распространение развитых ранее теории изотопозамещения и «расширенного метода локальных мод» на несимметрично - замещенные изотопические модификации аксиально-симметричных молекул и трехатомные молекулы с произвольной величиной угла между связями.
- Разработка и апробация на реальных спектрах высокого разрешения метода «SPGF» («спектроскопический потенциал - глобальный фиттинг»).
- Модернизация модели эффективных вращательных операторов с целью расширения области ее применения к системам, включающим в себя до нескольких десятков сильно взаимодействующих колебательно – вращательных полос и ее апробация на дейтероизвод-ных модификациях молекул H2O и CH4.
- Для молекул высокой симметрии разработка и реализация на основе использования операторной теории возмущений, теории неприводимых тензорных операторов и языков аналитического программирования MAPLE и MATHEMATIKA подхода, позволяющего определять аналитические формулы для различных спектроскопических параметров в виде функций равновесных структурных параметров и параметров ВМПФ молекулы.
- Разработка и практическая реализация метода определения вращательной структуры основного колебательного состояния молекул типа асимметричного волчка симметрии CS (известен в настоящее время как «метод двух пар переходов»).
- Интерпретация и дальнейший теоретический анализ спектров высокого разрешения трех-, четырех-, пяти-атомных молекул различной симметрии с целью извлечения из них информации, необходимой для последующего решения задачи определения внутримолекулярной потенциальной функции молекул.
Основные методы исследования. Исходя из перечисленных задач, для их решения применялись методы теории неприводимых тензорных операторов, методы операторной теории возмущений, вычислительные методы с использованием языков программирования FORTRAN, MAPLE и MATHEMATIKA. В силу специфики задач, широко использовались как традиционные, так и разработанные с участием автора методы теоретической спектроскопии, в частности теория колебательно-вращательных взаимодействий в многоатомных молекулах. Экспериментальные исследования, которые, с одной стороны, необходимо было выполнять для решения основной задачи данной работы, и которые, с другой стороны, необходимы для апробации корректности основных результатов и выводов работы, основаны на методах лазерной и Фурье-спектроскопии высокого разрешения.
Научные положения, выносимые на защиту:
- Развитый в работе и основанный на использовании трансформационных свойств коле-бательных координат и методов операторной теории возмущений метод определения внутримолекулярной потенциальной функции существенно проще в практических приложениях по сравнению с аналогичными известными методами. Вместе с тем он позволяет определять параметры потенциальной функции с точностью не хуже чем точности известных полуэмпирических методов и применим к любым многоатомным нормальным молекулам.
- Метод "глобального фиттинга", основанный на возможности определять зависимость всех спектроскопических параметров модели от колебательных квантовых чисел и развитый в работе применительно к трехатомным молекулам симметрии Cs, дает возможность описывать весь колебательно - вращательный спектр нелинейных молекул типа XYZ.
- Развитый и реализованный на примере трехатомных молекул типа асимметричного волчка SPGF-метод (Spectroscopic Potential - Global Fit), сочетая в себе простоту и эффективность ранее развитых метода "глобального фиттинга" и метода определения ВМПФ молекулы, позволяет корректно описывать всю колебательно - вращательную структуру одновременно для всех изотопомеров молекулы.
- Принадлежность молекулы XH3 к классу молекул, удовлетворяющих условиям модели локальных мод, приводит к наличию у низкосимметричных изотопических модификаций XH2D и XHD2 специфических особенностей. А именно, при исследовании валентно возбужденных состояний таких молекул их фрагменты XH2/XD2 и XD/XH проявляют спектроскопические свойства, присущие независимым двухатомной XD/XH и трехатомной "локально-модной" XH2/XD2 молекулам.
- Наличие простых аналитических соотношений между константами форм колебаний различных изотопических модификаций молекулы, удовлетворяющей "расширенной модели локальных мод", позволяет устанавливать в аналитическом виде зависимости различных спектроскопических параметров низко-симметричных изотопомеров молекулы от экспериментальных значений спектроскопических параметров ее более симметричной «материнской модификации».
Достоверность результатов и выводов диссертационной работы обеспечивается строгостью используемых математических методов и моделей, непротиворечивостью результатов и выводов, их согласованностью с современными представлениями теоретической колебательно – вращательной спектроскопии и, там где было возможно, результатами других авторов.
- Вновь разработанные и/или усовершенствованные методы интерпретации спектров высокого разрешения обеспечивают качество их обработки на уровне погрешностей современного эксперимента инфракрасного и видимого диапазонов;
- Выполняемые на основе результатов решения обратных спектроскопических задач расчеты предсказательного характера положений линий исследуемых молекул показывают вполне удовлетворительную точность, достаточную для безальтернативной интерпретации спектров высокого разрешения.
- Получаемые на основе разработанного метода определения ВМПФ молекулы параметры потенциальных функций различных молекул позволяют восстанавливать и предсказывать центры колебательно – вращательных полос, основные вращательные и резонансные параметры с точностью, по меньшей мере, не хуже чем любой из известных на настоящий момент методов.
Научная новизна работы определяется, в первую очередь, разработанным простым и, вместе с тем, эффективным методом определения внутримолекулярной потенциальной функции произвольной многоатомной молекулы, Следует также отметить, что
- впервые в практике колебательно – вращательной спектроскопии в рамках разработанного метода глобального «фитинга» выполнен совместный анализ 22 полос (в совокупности более 9700 колебательно – вращательных линий) молекулы D2S и 20 полос (около 10400 линий) молекулы HDS;
- впервые зарегистрирован при температуре 78 К Фурье-спектр высокого разрешения молекул CH2D2, CH3D и CHD3, выполнен его теоретический анализ и на этой основе определены с точностью 1- 2 х 10-4 см-1 центры и вращательная структура около 200-т ранее неизвестных полос этих трех дейтеропроизводных модификаций метана (ранее была известна информация лишь о 37 полосах этих молекул);
- на этой основе впервые с точностью достаточной для корректного предсказания вращательных, центробежных и резонансных параметров дейтеропроизводных модификаций определена ВМПФ молекулы метана и на этой основе
- впервые в практике спектроскопии выполнен совместный анализ представляющих собой квазиконтинуум 22-х и 89-и сильно взаимодействующих колебательно – вращательных полос молекулы CH2D2;
- впервые в рамках разработанного автором и реализованного в виде комплекса программ SPGF-метода выполнен совместный анализ всех известных на настоящий момент экспериментальных данных для молекул H2S, D2S и HDS, имеющий, с одной стороны, самостоятельное важное значение для дальнейшего развития спектроскопии и, с другой стороны, создавший основу для количественного определения параметров ВМПФ молекулы сероводорода;
- впервые в рамках разработанного метода определения равновесных структурных параметров молекулы определены на основе только экспериментальных данных и без привлечения информации о резонансных характеристиках молекулы фосфина ее равновесный угол и длина валентной связи;
- впервые в молекулах AsD3 и PD3 экспериментально зарегистрированы и теоретически описаны а1/а2 расщепления в уровнях со значением квантового числа K = 4, 5 и 7, а в молекуле ; PH3 – вплоть до K = 10;
- в результате компиляции положений и результатов теории изотопозамещения и «расширенного метода локальных мод» получен ряд новых ранее неизвестных изотопических соотношений между различными спектроскопическими параметрами симметричных и несимметричных изотопических модификаций четырехатомных молекул пирамидального типа;
- на основе анализа экспериментальных данных показано, что в молекулах ХH2D/ XHD2 (X = P, As) фрагменты ХH/ XD и ХH2/XD2 с достаточно высокой степенью достоверности могут рассматриваться как независимые и, при этом, фрагмент ХH2/XD2 со спектроскопической точки зрения ведет себя подобно молекуле ХH2, удовлетворяющей модели локальных мод.
- выполнен теоретический анализ в общей сложности более чем 400 впервые зарегистрированных с высоким разрешением колебательно – вращательных полос (более 80000 линий поглощения, зарегистрированных с точностями в центах поглощения на уровне 1- 5 х 10-4 см-1 ) молекул H2S, D2S, HDS, H2Se, D2O, HDO, PH3, PH2D, PHD2, PD3, AsH2D, AsHD2, AsD3, D2CO, CH2D2, CH3D, CHD3.
Научная ценность положений и полученных результатов определяется следующим:
- Разработанный метод определения внутримолекулярной потенциальной функции на основе экспериментальных данных о ее центрах полос и колебательно – вращательных энергиях, с одной стороны, является существенно более простым в реализации по сравнению с любым известным в настоящее время аналогичным методом и, в то же время, может быть применен к произвольной нормальной молекуле;
- Развитая и апробированная на четырехатомных аксиально симметричных молекулах теория изотопозамещения в молекулах удовлетворяющих модели локальных мод дает рецепт и позволяет получить многочисленные ранее неизвестные изотопические соотношения между различными спектроскопическими параметрами молекул;
- Предложенный в работе метод определения равновесных структурных параметров молекулы позволяет определять с высокой точностью равновесную структуру многоатомных молекул на основе только экспериментальной информации о положениях колебательно – вращательных линий и без привлечения дополнительной информации о характере и силе резонансных взаимодействий;
- Развитые в работе методы позволяют однозначно разрешить проблемы, традиционно возникающие в молекулярной спектроскопии при исследовании спектров сильно взаимодействующих колебательно – вращательных полос различного типа молекул и связанные с неоднозначностью и сильной корреляцией спектроскопических параметров в резонансных и диагональных блоках эффективных вращательных операторов.
Практическая значимость результатов заключается, прежде всего в том, что разработанные в процессе выполнения работы модели и методы, результаты применения развитых методов и моделей к исследованию реальных объектов позволяют получить количественную информацию о параметрах спектральных линий и фундаментальных характеристиках молекул различного типа, которая является необходимой при исследовании более сложных эффектов внутримолекулярной природы, оптических и физико – химических свойств молекул, является важной для пополнения баз данных и банков спектроскопической информации, используемой в астрофизике, газоанализе, атмосферной оптике и т.д.
Связь с плановыми работами. Работа выполнялась в рамках плановых научно – исследовательских работ Томского государственного университета по программам
- «Разработка средств контроля малых газовых примесей методами СВЧ и лазерной спектроскопии», №1.28.96.Ф.
- «Исследование фотофизических т фотохимических свойств молекул», №1.4.01.
Часть работ была выполнена при финансовой поддержке программы «Университеты России 2000-2001»; по грантам Министерства образования Российской федерации №97-5.1-7 МО, №97-5.1-8 МО, №Е00-3.2-192 МО, №Э-00-2.0-19, №97-9.3-17 МО; индивидуальным грантам Министерства образования Российской федерации для выполнения совместных научных исследований в зарубежных научных центрах (Хефей, КНР, 2001 и Дижон, Франция, 2003); гранту Президента Российской федерации № МК-4740.2007.2.
Внедрение результатов диссертации и рекомендации по их дальнейшему использованию. Результаты по теме диссертации использовались при выполнении совместных научных исследований Томского госуниверситета и университетов Оулу (Финляндия), Вупперталя (Германия), Парижа, Дижона и Лилля (Франция), Хефея и Шанхая (Китай), Болоньи (Италия) и Цюриха (Швейцария). Многочисленная высокоточная спектроскопическая информация, полученная в процессе выполнения работы может быть использована в организациях, занимающихся исследованиями в области физики молекул, молекулярной спектроскопии, атмосферной оптики, физики газовых сред, астрофизики, планетологии и др., в частности, Институт оптики атмосферы СО РАН (г. Томск), Институт прикладной физики РАН (г. Нижний Новгород), Институт спектроскопии РАН (г. Троицк, Московская обл.), Институт общей физики РАН (г. Москва), Московский и Санкт-Петербургский госуниверситеты. Значительная часть представленных в диссертационной работе результатов используется при чтении курсов лекций «Современные проблемы молекулярной спектроскопии» и «Физика атомов и молекул» в Томском государственном университете.
Апробация работы. Основные результаты диссертационной работы докладывались автором на XXV Европейском конгрессе молекулярной спектроскопии (Коимбра, Португалия, 2000); 16-ой международной конференции по молекулярной спектроскопии высокого разрешения (Прага, Чехия, 2000); 17-ом международном коллоквиуме по молекулярной спектроскопии высокого разрешения (Арнем, Голландия, 2001); 17-ой международной конференции по молекулярной спектроскопии высокого разрешения (Прага, Чехия, 2002); 18-ом международном коллоквиуме по молекулярной спектроскопии высокого разрешения (Дижон, Франция, 2003); 39-ом международном симпозиуме по теоретической химии (Гватт, оз. Тюн, Швейцария, 2003); 19-ом международном коллоквиуме по молекулярной спектроскопии высокого разрешения (Саламанка, Испания, 2005); 19-ой международной конференции по молекулярной спектроскопии высокого разрешения (Прага, Чехия, 2006); 61-ом международном симпозиуме по молекулярной спектроскопии (Колумбус, Огайо, США, 2006); 20-ом международном коллоквиуме по молекулярной спектроскопии высокого разрешения (Дижон, Франция, 2007). Отдельные проблемы, затронутые в диссертации, многократно докладывались автором в период 2001 – 2007 г.г. на научных семинарах лаборатории физики университета Бургундии (Дижон, Франция), лаборатории фотофизики молекул университета Париж-юг (Орсэ, Франция), отдела неорганической химии университета Вупперталя (Вупперталь, Германия), физическом факультете университета Оулу (Оулу, Финляндия), Национальной лаборатории физических исследований университета наук и технологий Китая (Хефей, Китай), лаборатории физической химии Федерального института высоких технологий Швейцарии (Цюрих, Швейцария).
Публикации. Материалы диссертации в полном объеме опубликованы в научной печати (всего 87 публикаций), в том числе в специализированных международных рецензируемых журналах (38 статей), в рецензируемых отечественных журналах (6 статей), известиях Томского политехнического университета (2 статьи), депонированных в ВИНИТИ (2 статьи), тезисах и трудах международных конференций (39 публикаций).
Вклад автора. Основные результаты диссертационной работы получены лично автором как в индивидуальных, так и в коллективных исследованиях. Вклад автора на разных этапах выражался в постановке решаемых задач, разработке путей и методов их решения, разработке алгоритмов и создании программ расчета на языках FORTRAN и MAPLE, проведении непосредственных расчетов, обсуждении и интерпретации полученных в ходе выполнения работ результатов. Постановка задач и выбор путей их решения осуществлялись совместно с научным консультантом, профессором О. Н. Улениковым. Часть исследований (в частности, разработка и практическая реализация в виде алгоритмов и программ метода «глобального фиттинга», а также совместный анализ на этой основе колебательно – вращательных спектров молекул D2S и HDS) выполнены совместно с Г. А. Онопенко. Исследования по разработке первых модификаций развитого в диссертации метода определения внутримолекулярной потенциальной функции многоатомных молекул были выполнены совместно с Е. А. Синицыным, научным руководителем кандидатской диссертации которого была автор данной работы. Совместно с Е. А. Синицыным и Г. А. Онопенко были также выполнены работы по реализации и апробации оригинального метода определения равновесной структуры молекул и получены новые результаты в теории изотопозамещения молекул, удовлетворяющих модели локальных мод. Совместно с Ю. Б. Юхник (научным руководителем кандидатской диссертации которой также была автор данной работы) выполнены теоретические исследования тонкой вращательной структуры аксиально-симметричных молекул PD3 и AsD3 и получены первые предварительные оценки параметров ВМПФ молекул фосфина и арсина. Совместно с аспиранткой О. В. Громовой проведено исследование в рамках модели эффективных операторов спектров высокого разрешения различных изотопических модификаций молекулы сероводорода. Определенный вклад на уровне постановки задачи и выборе путей ее реализации внесен автором данной работы в решение задачи оценки параметров ВМПФ аксиальносимметричных молекул типа XH3 алгебраическими методами, которая выполнялась совместно с Н. А. Санжаровым и К. Леруа. Значительное число работ, результаты которых вошли в данную диссертацию, выполнено в соавторстве с зарубежными учеными, вклад которых заключался в выполнении экспериментальной части исследований. Вклад автора диссертации в экспериментальные исследования ограничивался участием в постановке задачи и выдаче рекомендаций для реализации оптимальных условий эксперимента.
Объем и структура диссертации. Диссертация состоит из введения, 6 глав, заключения и списка литературы, включающего 269 наименований. Диссертация содержит 167 страниц текста плюс 86 таблиц и 36 рисунков.
КРАТКОЕ Содержание работы
Во введении обоснована актуальность темы, кратко изложены предмет исследований и структура диссертации, сформулированы цели работы, защищаемые положения, научная и практическая значимость работы.
Первая глава носит обзорный характер и содержит краткое описание необходимых для понимания оригинальной части работы принципов и некоторых методов колебательно - вращательной теории, включающих способ построения во внутримолекулярных координатах квантово - механического гамильтониана для произвольной многоатомной молекулы; метод решения колебательно - вращательной задачи с использованием эффективных вращательных операторов, в том числе, в симметризованной форме; основные сведения из теории изотопозамещения в многоатомных молекулах и «расширенного метода локальных мод; последний параграф первой главы посвящён теории неприводимых тензорных операторов в том объёме, который необходим для понимания результатов решения задачи по определению ВМПФ молекул на основе экспериментальной информации о спектрах молекул аксиальной симметрии или аксиально - симметричных изотопомеров молекул сферической симметрии.
Вторая глава посвящена изложению вопросов, связанных с решением основной задачи диссертационной работы - разработке простого и, вместе с тем, эффективного метода определения ВМПФ произвольной многоатомной нормальной молекулы на основе экспериментальной информации о ее спектрах высокого разрешения. Следует заметить, что корректное решение данной задачи, представляя, в сущности, неограниченные возможности применения ее результатов в различных областях как чисто академических, так и прикладных наук, требует для своей реализации тщательного исследования целого комплекса проблем. Среди них следует отметить проблему построения корректного и, вместе с тем, максимально простого для дальнейшего использования гамильтониана молекулы, который можно было бы без изменений (или, по крайней мере, с минимальными изменениями) применять для описания спектров любых многоатомных молекул; нетривиальную в общем случае проблему построения матрицы гамильтониана, в особенности для молекул высокой симметрии; проблему многократной диагонализации матриц огромной размерности, что существенно ограничивает область применимости подавляющего большинства использующихся в настоящее время для решения аналогичных задач методов молекулами с малым числом атомов.
В связи со сказанным во второй главе диссертации рассмотрены различные аспекты разработанного нами метода определения ВМПФ многоатомных молекул. Как показывает анализ подобного рода задач, чрезвычайно важным для их успешного решения является проблема удачного выбора координат, описывающих смещения ядер в молекуле друг относительно друга. Такими координатами могут быть, в частности, аналоги нормальных координат ![]() , естественные координаты
, естественные координаты ![]() и
и ![]() , координаты симметрии
, координаты симметрии ![]() , координаты Морзе
, координаты Морзе ![]() и другие. Важно заметить, что последние три типа колебательных координат являются изотопически инвариантными, в то время как координаты типа
и другие. Важно заметить, что последние три типа колебательных координат являются изотопически инвариантными, в то время как координаты типа ![]() связаны только с конкретным изотопомером молекулы.
связаны только с конкретным изотопомером молекулы.
Следует отметить, что в силу принципиальной невозможности прямого решения задачи определения ВМПФ даже для самых простых молекул, любой метод представляет собой ту или иную итерационную процедуру. Как следствие, возникает вопрос о начальном приближении для тех величин, которые являются результатом решения задачи. В нашем случае, такими величинами являются равновесные структурные параметры и параметры ВМПФ. Наличие достаточно хорошего начального приближения для параметров итерационной процедуры позволяет определить так называемые константы форм колебаний ![]() , имеющих фундаментальное значение как для колебательно - вращательной теории в целом, так и для решения основной задачи данной диссертации (практически все величины в колебательно – вращательной теории в той или иной степени зависят от констант форм колебаний). Поэтому в первом параграфе второй главы описана реализованная нами итерационная расчетная схема решения систем нелинейных уравнений (которых для молекул с большим числом атомов может быть до нескольких сотен), позволяющая численно определить константы форм колебаний для любой многоатомной нормальной молекулы, независимо от количества ядер и симметрии молекулы.
, имеющих фундаментальное значение как для колебательно - вращательной теории в целом, так и для решения основной задачи данной диссертации (практически все величины в колебательно – вращательной теории в той или иной степени зависят от констант форм колебаний). Поэтому в первом параграфе второй главы описана реализованная нами итерационная расчетная схема решения систем нелинейных уравнений (которых для молекул с большим числом атомов может быть до нескольких сотен), позволяющая численно определить константы форм колебаний для любой многоатомной нормальной молекулы, независимо от количества ядер и симметрии молекулы.
В параграфе 2.2 второй главы на основе результатов параграфа 1.1 определены связи между колебательными координатами различного типа, необходимые для дальнейшего анализа. В качестве иллюстрации ниже приведены связи между координатами ![]() и естественными координатами
и естественными координатами ![]() и
и ![]() (изменения длин валентных связей и углов между валентными связями. Поскольку в работе речь идет о нормальных молекулах, то результаты получены в виде рядов
(изменения длин валентных связей и углов между валентными связями. Поскольку в работе речь идет о нормальных молекулах, то результаты получены в виде рядов
![]() (1)
(1)
![]() (2)
(2)
Входящие в разложения коэффициенты ![]() и
и ![]() определены в виде аналитических функций масс ядер молекулы, ее равновесных структурных параметров и определенных в параграфе 2.1 констант форм колебаний
определены в виде аналитических функций масс ядер молекулы, ее равновесных структурных параметров и определенных в параграфе 2.1 констант форм колебаний ![]() .
.
В соответствие с общими принципами колебательно – вращательной теории, точный гамильтониан нормальной молекулы (далее мы будем пока говорить о колебательном гамильтониане) в приближении Борна – Оппенгеймера может быть представлен в виде
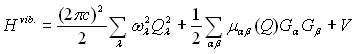 , (3)
, (3)
где
![]() . (4) Важно заметить, что первые два слагаемых в (3) являются точным оператором кинетической энергии, зависящим от координат
. (4) Важно заметить, что первые два слагаемых в (3) являются точным оператором кинетической энергии, зависящим от координат ![]() , и справедливы для любой нормальной молекулы; потенциальную же функцию
, и справедливы для любой нормальной молекулы; потенциальную же функцию ![]() в (3) разумно использовать в виде функции изотопически инвариантных координат. Как следствие, для дальнейшего решения задачи возникает проблема перехода от координат
в (3) разумно использовать в виде функции изотопически инвариантных координат. Как следствие, для дальнейшего решения задачи возникает проблема перехода от координат ![]() к изотопически независимым координатам потенциальной функции
к изотопически независимым координатам потенциальной функции ![]() , или наоборот.
, или наоборот.
В принципе, преобразования могут быть осуществлены в обоих направлениях. Однако, как показал проведенный нами анализ, переход к любому из трех вышеупомянутых типов изотопически инвариантных координат в кинетической части оператора (3) приводит к чрезвычайно сложным выражениям. Поэтому, в параграфе 2.3 описан подход, позволяющий сохранить кинетическую часть гамильтониана молекулы в весьма простом виде. С целью упрощения понимания сути выполняемых преобразований рассмотрение ведется на примере трехатомной нелинейной молекулы XY2 симметрии C2v. Минимальные изменения в общем подходе, которые необходимо выполнить в случае иных изотопических модификаций молекулы или молекул с большим числом атомов и/или другой симметрии рассматриваются в последующих главах диссертации при применении общего подхода к конкретным молекулам.
В колебательно – вращательной теории известно, что одним из наиболее трудноразрешимых вопросов, возникающих при использовании удобных во многих других отношениях нормальных координат, являются расходимости вызванные наличием в потенциальной функции молекулы параметров ![]() , ассоциированных с валентными колебаниями. В то же время, использование в качестве переменных координат Морзе приводит к такой формулировке задачи, которая полностью свободна от указанной проблемы. Как следствие, основная идея построения корректного, но простого для дальнейшего использования гамильтониана молекулы, заключается в переходе в точном операторе (3) к такому набору координат, которые бы, с одной стороны, были координатами Морзе-типа для валентных колебаний, но, вместе с тем, позволили бы сохранить кинетическую часть гамильтониана в простом виде. Описанные в параграфе 2.3 преобразования позволили получить точный гамильтониан (3) молекулы в чрезвычайно простом виде
, ассоциированных с валентными колебаниями. В то же время, использование в качестве переменных координат Морзе приводит к такой формулировке задачи, которая полностью свободна от указанной проблемы. Как следствие, основная идея построения корректного, но простого для дальнейшего использования гамильтониана молекулы, заключается в переходе в точном операторе (3) к такому набору координат, которые бы, с одной стороны, были координатами Морзе-типа для валентных колебаний, но, вместе с тем, позволили бы сохранить кинетическую часть гамильтониана в простом виде. Описанные в параграфе 2.3 преобразования позволили получить точный гамильтониан (3) молекулы в чрезвычайно простом виде
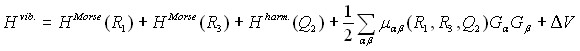 , (5)
, (5)
где
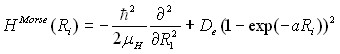 (6)
(6)
-осцилляторы Морзе для координат ![]() , ассоциированных с валентными связями;
, ассоциированных с валентными связями; ![]() – оператор традиционного гармонического осциллятора для координаты
– оператор традиционного гармонического осциллятора для координаты ![]() , связанной с изменением валентного угла; потенциальная функция
, связанной с изменением валентного угла; потенциальная функция ![]() в самой общей форме (без учета вкладов от потенциальной функции в
в самой общей форме (без учета вкладов от потенциальной функции в ![]() и
и ![]() ) имеет вид
) имеет вид
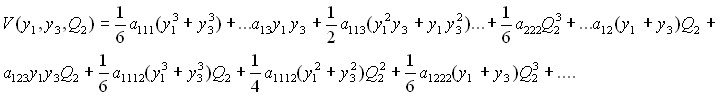 (7)
(7)
Четвертое слагаемое в (5) представляет собой в данном подходе весьма простой результат преобразования оператора ![]() из (3). Важно отметить особенности полученного результата: (а). гамильтониан (5) - (7) является точным и в результате минимальных модификаций может быть применен к любой изотопической модификации любой многоатомной нормальной молекулы; (б). для молекул с малым числом атомов он является аналогом ЕКЕ операторов, но существенно превосходит их по простоте (для молекул с большим числом атомов соответствующие EKE операторы отсутствуют). В качестве иллюстрации эффективности и корректности полученного результата оператор в форме (5) – (7) был применен для определения параметров потенциальной функции молекулы H2S. В качестве исходных «экспериментальных» данных был взят 141 центр полос с
из (3). Важно отметить особенности полученного результата: (а). гамильтониан (5) - (7) является точным и в результате минимальных модификаций может быть применен к любой изотопической модификации любой многоатомной нормальной молекулы; (б). для молекул с малым числом атомов он является аналогом ЕКЕ операторов, но существенно превосходит их по простоте (для молекул с большим числом атомов соответствующие EKE операторы отсутствуют). В качестве иллюстрации эффективности и корректности полученного результата оператор в форме (5) – (7) был применен для определения параметров потенциальной функции молекулы H2S. В качестве исходных «экспериментальных» данных был взят 141 центр полос с ![]() , рассчитанных в (Halonen, Carrington, J. Chem, Phys., 88, 4171 (1988)) в рамках существенно более сложной модели по сравнению с нашей. С использованием нашей модели все центры полос были воспроизведены со средней точностью 0.69 см-1 только шестью параметрами. Аналогичный расчет для молекулы D2S в рамках развитой модели позволил воспроизвести шестью параметрами 141 центр полос из той же работы со средней точностью 0.56 см-1.
, рассчитанных в (Halonen, Carrington, J. Chem, Phys., 88, 4171 (1988)) в рамках существенно более сложной модели по сравнению с нашей. С использованием нашей модели все центры полос были воспроизведены со средней точностью 0.69 см-1 только шестью параметрами. Аналогичный расчет для молекулы D2S в рамках развитой модели позволил воспроизвести шестью параметрами 141 центр полос из той же работы со средней точностью 0.56 см-1.
Введенные выше координаты ![]() не являются изотопически инвариантными. Поэтому для возможности использования полученной модели для одновременного рассмотрения нескольких изотопических модификаций ее следует несколько модифицировать. Как показано в параграфе 2.4, проблема может быть решена на основе результатов, полученных в 2.1 и 2.2, если потенциальную функцию в исходном выражении (3) взять в виде функции естественных координат молекулы
не являются изотопически инвариантными. Поэтому для возможности использования полученной модели для одновременного рассмотрения нескольких изотопических модификаций ее следует несколько модифицировать. Как показано в параграфе 2.4, проблема может быть решена на основе результатов, полученных в 2.1 и 2.2, если потенциальную функцию в исходном выражении (3) взять в виде функции естественных координат молекулы ![]() и
и ![]() . При этом кинетическая часть гамильтониана молекулы остается такой же простой, как и прежде, а потенциальная функция для каждой изотопической модификации молекулы преобразуется к уже известному выражению (7) с той лишь разницей, что каждый из коэффициентов
. При этом кинетическая часть гамильтониана молекулы остается такой же простой, как и прежде, а потенциальная функция для каждой изотопической модификации молекулы преобразуется к уже известному выражению (7) с той лишь разницей, что каждый из коэффициентов ![]() зависит теперь известным образом от параметров
зависит теперь известным образом от параметров ![]() потенциальной функции
потенциальной функции ![]() . Это обстоятельство позволяет в рамках одной модели с использованием единой потенциальной функции проводить исследование спектров всех без исключения изотопических разновидностей молекулы. Вторым важным следствием полученного результата является то, что метод дает возможность использовать одновременно экспериментальные данные для всех без исключения изотопических модификаций молекулы, что существенно повышает устойчивость решения и достоверность результатов.
. Это обстоятельство позволяет в рамках одной модели с использованием единой потенциальной функции проводить исследование спектров всех без исключения изотопических разновидностей молекулы. Вторым важным следствием полученного результата является то, что метод дает возможность использовать одновременно экспериментальные данные для всех без исключения изотопических модификаций молекулы, что существенно повышает устойчивость решения и достоверность результатов.
Параграф 2.5 диссертации посвящен решению второй проблемы, традиционно возникающей при использовании современных вариационных методов решения задачи определения ВМПФ многоатомной молекулы. Проблема заключается в том, что при использовании любого подобного метода возникает необходимость многократного построения и последующей диагонализации матрицы гамильтониана. При этом даже использование существенно более простого по сравнению с традиционными гамильтониана молекулы приводит к необходимости работать с матрицами огромной размерности. Эта проблема многократно обсуждалась в литературе, однако до сих пор оставалась нерешенной, что являлось вторым серьезным ограничением области применимости используемых вариационных методов молекулами с малым числом атомов.
Нами, по аналогии с известной операторной теорией возмущений, был разработан и реализован в виде алгоритма и программ на языке FORTRAN метод определения собственных значений гамильтониана молекулы, который позволяет в значительной степени избежать проблем, характерных для традиционных методов. Суть разработанного нами подхода сводится не к построению матрицы гамильтониана большой размерности, а к анализу по описанной в параграфе 2.5 схеме определенных совокупностей матричных элементов гамильтониана из разделов 2.3 – 2.4. В результате такого анализа задача сводится к диагонализации матриц приемлемой размерности (от 50 до 1000 строк и столбцов для изотопомеров пятиатомной молекулы метана).
Расчетная схема представляет собой вариант теории возмущений. В разделе 2.5 сформулированы критерии возможности редукции матриц большой размерности к матрицам существенно меньших размерностей. В качестве иллюстрации, один из критериев представлен ниже
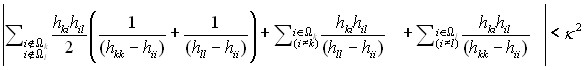 , (8)
, (8)
В качестве теста корректности разработанного подхода нами были выполнены расчеты собственных значений гамильтониана в форме (5) – (7) для различных комбинаций параметров ![]() . На первом этапе строилась и напрямую диагонализировалась достаточно большая матрица, вплоть до 10000 строк и столбцов. На втором этапе к такой матрице применялась описанная выше процедура. Как показало сравнение, разница между двумя расчетами не превышает величин порядка 0.4 – 0.6 см-1, что является вполне приемлемой точностью, если иметь в виду что в данной диссертации рассмотрение ведется в рамках приближения Борна – Оппенгеймера.
. На первом этапе строилась и напрямую диагонализировалась достаточно большая матрица, вплоть до 10000 строк и столбцов. На втором этапе к такой матрице применялась описанная выше процедура. Как показало сравнение, разница между двумя расчетами не превышает величин порядка 0.4 – 0.6 см-1, что является вполне приемлемой точностью, если иметь в виду что в данной диссертации рассмотрение ведется в рамках приближения Борна – Оппенгеймера.
В последующих разделах диссертации разработанный и описанный выше метод используется для определения ВМПФ реальных 3 - 5 атомных молекул различной симметрии. При этом, как уже отмечалось выше, вторым весьма важным условием возможности реализации разработанного метода является комплекс проблем, связанных с обеспечением задачи определения ВМПФ исходной экспериментальной информацией. В качестве такого рода экспериментальной информации могут использоваться как положения колебательно – вращательных линий исследуемой молекулы, так и различного рода промежуточные результаты, полученные в результате решения обратных спектроскопических задач (далее ОСЗ) на основе использования все тех же положений колебательно – вращательных линий молекулы. При этом, как показывает анализ состояния дел в современной колебательно–вращательной спектроскопии, для успешного и корректного решение ОСЗ для различного типа молекул во многих случаях необходимо не только модифицировать уже известные, но и разрабатывать новые методы и подходы. Ряд таких подходов и методов, разработанных автором или при непосредственном его участии рассматривается далее в разделах, посвященных исследованию конкретных молекул.
Третья глава диссертации посвящена рассмотрению очень схожих друг с другом со спектроскопической точки зрения молекул сероводорода и селеноводорода. При этом, основное внимание уделено рассмотрению вопросов, связанных с исследованием спектров высокого разрешения молекулы H2S и ее дейтеро- и дважды дейтеро-замещенных модификаций и использованию полученных результатов в задаче определения ВМПФ сероводорода на основе развитого в главе 2 метода. Что касается молекулы H2Se, ее спектры в данной диссертации не исследуются. Основной интерес к этой молекуле в диссертации объясняется тем фактом, что ранее автором диссертации совместно с Йенсеном, Улениковым и Санжаровым была выполнена работа по определению потенциальной функции молекулы H2Se методом MORBID. Имея в виду потребность в огромных компьютерных ресурсах (память и время счета) для реализации MORBID процедуры, представляется интересным выполнить сравнительные расчеты параметров потенциальной функции и разработанным нами методом.
Заметим, что исследование молекулы сероводорода представляет большой как чисто академический (молекула H2S является одной из наиболее легких молекул, поэтому все эффекты и особенности, присущие молекулам типа асимметричного волчка, наиболее ярко проявляются в их спектрах), так и прикладной интерес. С точки же зрения обеспечения экспериментальной информацией задачи определения ВМПФ сероводорода, интерес представляют результаты исследования не только спектров основной изотопической модификации H2S, но и D2S и, в особенности, HDS. Последнее обусловлено известным фактом, что экспериментальная информация о спектрах одной изотопической модификации позволяет определить лишь отдельные из параметров потенциальной функции.
В параграфе 3.1 приведены результаты обширных исследований спектров высокого разрешения молекулы H2S в районах 5000 – 6700, 7300 – 7900 и 8500 – 8900 см-1. Зарегистрированные на Фурье-спектрометре Bruker IFS 120 HR в университете наук и технологий Китая (Хефей, КНР), колебательно–вращательные спектры трех полиад взаимо-действующих колебательных состояний (полосы ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() , содержащие около 4000 линий поглощения) были исследованы в рамках традиционной для колебательно – вращательной теории модели эффективных операторов, которая хорошо работает применительно к такого рода совокупностям полос. Поэтому, мы не будем останавливаться подробно на интерпретации зарегистрированных спектров и последующем решении на этой основе необходимых обратных спектроскопических задач. Заметим лишь, что полученные результаты представляют большой самостоятельный интерес для многих задач физики и химии, в которых необходимы данные о параметрах спектральных линий молекулы H2S. Для нас же полученные в данном разделе результаты представляют интерес в качестве высокоточной исходной экспериментальной информации в задаче определения ВМПФ молекулы сероводорода. Отметим также, что при исследовании спектра высокого разрешения был экспериментально зарегистрирован и количественно описан эффект полного «заимствования интенсивности» (total borrowing of intensity), приводящий к исчезновению одной из линий дублетов в Р-ветви полос
, содержащие около 4000 линий поглощения) были исследованы в рамках традиционной для колебательно – вращательной теории модели эффективных операторов, которая хорошо работает применительно к такого рода совокупностям полос. Поэтому, мы не будем останавливаться подробно на интерпретации зарегистрированных спектров и последующем решении на этой основе необходимых обратных спектроскопических задач. Заметим лишь, что полученные результаты представляют большой самостоятельный интерес для многих задач физики и химии, в которых необходимы данные о параметрах спектральных линий молекулы H2S. Для нас же полученные в данном разделе результаты представляют интерес в качестве высокоточной исходной экспериментальной информации в задаче определения ВМПФ молекулы сероводорода. Отметим также, что при исследовании спектра высокого разрешения был экспериментально зарегистрирован и количественно описан эффект полного «заимствования интенсивности» (total borrowing of intensity), приводящий к исчезновению одной из линий дублетов в Р-ветви полос ![]() и
и ![]() , в то время как в R-ветви соответствующие дублеты-партнеры видны отчетливо.
, в то время как в R-ветви соответствующие дублеты-партнеры видны отчетливо.
Как отмечалось, основой для исследования спектров высокого разрешения молекулы H2S в предыдущем параграфе была модель эффективных операторов, широко использующаяся в современной колебательно – вращательной спектроскопии. Известно, однако, что эта модель обладает ограниченной областью применимости (параметры эффективного гамильтониана позволяют описывать вращательную структуру только одного или нескольких отдельных колебательных состояний) и, что более важно, не свободна от существенных недостатков, одним из которых является сильная корреляция между параметрами даже редуцированных эффективных гамильтонианов при решении обратных спектроскопических задач. Это, в свою очередь, приводит к тому, что параметры различных полиад, полученные из анализа экспериментальных данных, часто плохо согласуются друг с другом. Чтобы избежать указанных проблем, ранее в лаборатории молекулярной спектросокпии ТГУ был разработан метод «глобального фиттинга» применительно к трехатомным молекулам симметрии C2v. Особенностью этого метода является также то, что получаемые в его рамках параметры являются более фундаментальными по сравнению с параметрами эффективных операторов, поскольку они позволяют описывать вращательную структуру не отдельной совокупности колебательных полос, а весь колебательно – вращательный спектр молекулы. В следующем параграфе 3.2 метод «глобального фиттинга» используется для описания впервые зарегистрированного с участием автора спектра высокого разрешения молекулы D2S. Поскольку молекула D2S обладает симметрией C2v, то оказывается возможным использовать метод «глобального фиттинга» в ранее разработанной конфигурации. В то же время, подобный одновременный анализ 19-ти (из них 8 зарегистрировано впервые с участием автора) колебательно – вращательных полос молекулы HDS (симметрия Cs), потребовал модификации как теоретической основы метода, так и алгоритма расчета. Описание соответствующих результатов применительно к молекуле HDS составляет предмет параграфа 3.3. Ввиду громоздкости формул, описывающих модель, здесь для иллюстрации приведены лишь параметры одного из типов взаимодействий, описывающие резонансные взаимодействия типа Кориолиса между состояниями ![]() и
и ![]() :
:
![]() , (11)
, (11)
где ![]() ,
, ![]() ,
, ![]() ;
; ![]() - известные коэффициенты, связанные с различными комбинациями резонансных операторов углового момента
- известные коэффициенты, связанные с различными комбинациями резонансных операторов углового момента ![]() в части гамильтониана, описывающей резонанс Кориолиса выше обозначенного типа. Особо следует обратить внимание на наличие зависимости параметров
в части гамильтониана, описывающей резонанс Кориолиса выше обозначенного типа. Особо следует обратить внимание на наличие зависимости параметров ![]() модели от колебательных квантовых чисел, что и позволяет говорить о возможности описания данной моделью всего колебательно – вращательного спектра молекулы. Поскольку знание зависимости различных величин от колебательных квантовых чисел в данной модели является основополагающим, для корректного определения такой зависимости нами был разработан алгоритм, созданы программы и на этой основе выполнены специальные расчеты на языке аналитического программирования MAPLE c учетом высоких (вплоть до шестого) порядков операторной теории возмущений. Здесь следует отметить, по меньшей мере, еще два преимущества метода «глобального фиттинга» перед методом эффективных операторов: (а). он практически свободен от сильной корреляции основных параметров резонансных и диагональных блоков и (б). позволяет без каких бы то ни было проблем учитывать влияние так называемых «темных» полос. Статистическая информация по результатам совместного анализа, выполненного в рамках метода «глобального фиттинга», всех известных на настоящий день экспериментальных данных о положениях колебательно – вращательных линий молекул D2S и HDS приведена в параграфах 3.2 и 3.3. При этом учтены полосы как известные ранее, так и зарегистрированные впервые в рамках выполнения данной работы (в качестве иллюстрации один из зарегистрированных спектров, а именно, спектр молекулы HDS, приведен на рисунке 1). В параграфе 3.4 полученные для всех трех изотопических модификаций результаты используются в качестве исходной информации для решения задачи определения ВМПФ молекулы H2S на основе разработанного нами нового метода определения ВМПФ.
модели от колебательных квантовых чисел, что и позволяет говорить о возможности описания данной моделью всего колебательно – вращательного спектра молекулы. Поскольку знание зависимости различных величин от колебательных квантовых чисел в данной модели является основополагающим, для корректного определения такой зависимости нами был разработан алгоритм, созданы программы и на этой основе выполнены специальные расчеты на языке аналитического программирования MAPLE c учетом высоких (вплоть до шестого) порядков операторной теории возмущений. Здесь следует отметить, по меньшей мере, еще два преимущества метода «глобального фиттинга» перед методом эффективных операторов: (а). он практически свободен от сильной корреляции основных параметров резонансных и диагональных блоков и (б). позволяет без каких бы то ни было проблем учитывать влияние так называемых «темных» полос. Статистическая информация по результатам совместного анализа, выполненного в рамках метода «глобального фиттинга», всех известных на настоящий день экспериментальных данных о положениях колебательно – вращательных линий молекул D2S и HDS приведена в параграфах 3.2 и 3.3. При этом учтены полосы как известные ранее, так и зарегистрированные впервые в рамках выполнения данной работы (в качестве иллюстрации один из зарегистрированных спектров, а именно, спектр молекулы HDS, приведен на рисунке 1). В параграфе 3.4 полученные для всех трех изотопических модификаций результаты используются в качестве исходной информации для решения задачи определения ВМПФ молекулы H2S на основе разработанного нами нового метода определения ВМПФ.
Всего в качестве исходных данных были использованы центры 88 полос: 46 полос молекулы H2S (из них 14 определены из нашего исследования спектров молекулы H2S, см. параграф 3.1), 22 полосы молекулы D2S (из них 17 полос впервые с высоким разрешением исследовались нами, параграф 3.2) и 20 полос молекулы HDS
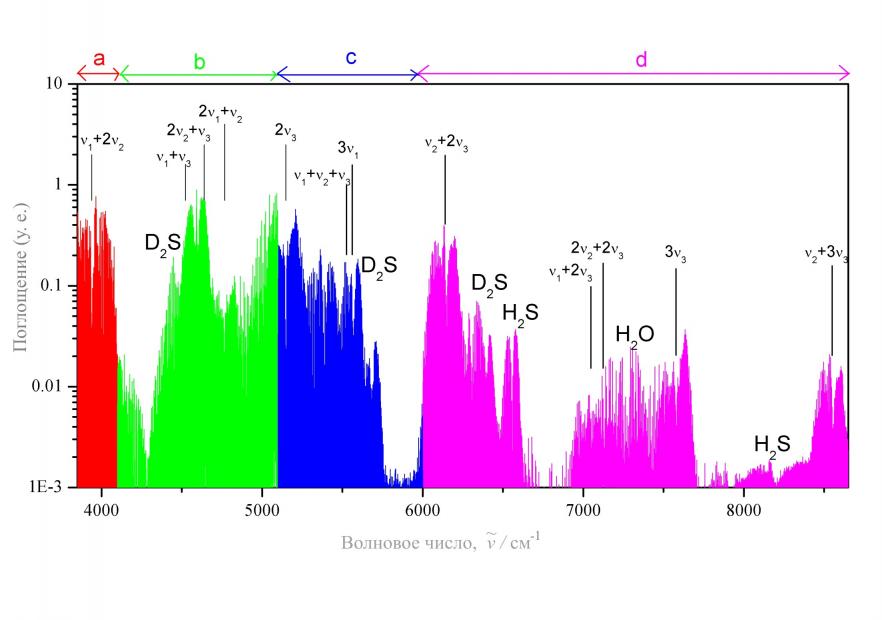
Рис. 1. Обзорный спектр молекулы HDS
(8 полос исследовались нами впер-вые, параграф 3.3). Заме-тим, что всего 16 параметров нашей модели воспроизводят положения 88 центров полос трех изотопии-ческих модификаций сероводо-рода со средней точностью 0.81 см-1, что является более чем удовлетворительным результатом, если учесть, что (а). рассмотрение в диссертации ведется в рамках приближения Борна – Оппенгеймера; (б). в исследование вовлечены и воспроизведены с более чем удовлетворительной точностью высоковозбужденные состояния с квантовыми числами валентных колебаний вплоть до ![]() и деформационного колебания вплоть до
и деформационного колебания вплоть до ![]() . Полученный результат, имея важное самостоятельное значение, является еще одним подтверждением эффективности развитого в данной работе метода определения ВМПФ молекул. Сравнение полученных нами результатов с лучшими на сегодняшний день более ранними работами (Halonen, Carrington, J. Chem, Phys., 88, 4171 (1988)) и (Kozin, Jensen. J. Mol. Spectrosc., 163, 483 (1994)) показывает, что полученный нами потенциал лучше описывает колебательную структуру изотопомеров сероводорода, чем результаты обеих цитированных работ, в особенности для состояний с высокими значениями квантовых. В частности, в первой из цитированных работ отклонения для некоторых из колебательных уровней достигают величин более 20 см-1, во второй (несмотря на то, что значения энергий в ней приведены для значений квантовых чисел валентных колебаний только для
. Полученный результат, имея важное самостоятельное значение, является еще одним подтверждением эффективности развитого в данной работе метода определения ВМПФ молекул. Сравнение полученных нами результатов с лучшими на сегодняшний день более ранними работами (Halonen, Carrington, J. Chem, Phys., 88, 4171 (1988)) и (Kozin, Jensen. J. Mol. Spectrosc., 163, 483 (1994)) показывает, что полученный нами потенциал лучше описывает колебательную структуру изотопомеров сероводорода, чем результаты обеих цитированных работ, в особенности для состояний с высокими значениями квантовых. В частности, в первой из цитированных работ отклонения для некоторых из колебательных уровней достигают величин более 20 см-1, во второй (несмотря на то, что значения энергий в ней приведены для значений квантовых чисел валентных колебаний только для ![]() ) – отклонения достигают величин более 5 см-1.
) – отклонения достигают величин более 5 см-1.
Как отмечалось выше, решение задачи описания спектров высокого разрешения молекул возможно на различном уровне общности. Мы уже упоминали традиционный метод эффективных операторов, который позволяет описывать вращательную структуру только отдельных совокупностей колебательных полос молекулы. Рассмотренный в данной главе метод «глобального фиттинга» представляет собой более высокий уровень описания спектров молекулы, поскольку его параметры дают возможность корректно описывать уже всю колебательно – вращательную структуру молекулы. В то же время, для описания колебательно – вращательных спектров иных изотопомеров молекулы даже в рамках метода «глобального фиттинга» требуется отдельный набор параметров. Наконец, самыми оптимальными являются параметры потенциальной функции, поскольку они позволяют описывать одним и тем же набором параметров колебательно – вращательные спектры всех без исключения изотопических модификаций молекулы. Иллюстрацией справедливости данного утверждения применительно к колебательной структуре служат результаты параграфа 3.4. Что касается колебательно – вращательных спектров, один из вариантов решения проблемы рассмотрен в разделе 3.5 третьей главы диссертации. Здесь рассматривается разработанный нами SPGF (Spectroscopic Potential – Global Fit) метод, практически реализованный на примере молекулы сероводорода. Метод представляет собой компиляцию развитого в главе 2 метода определения ВМПФ молекулы и метода «глобального фиттинга», описанного параграфах 3.2 – 3.3. Основу метода составляет модель, используемая в «глобальном фиттинге», но все наиболее значимые спектроскопические величины (центры полос и параметры различных колебательных резонансов, вращательные постоянные и основные колебательно – вращательные коэффициенты, основные параметры резонансов Кориолиса и основные центробежные коэффициенты) определяются в виде функций фундаментальных характеристик молекулы, то есть параметров потенциальной функции, задаваемой моделью, рассмотренной в главе 2 и разделе 3.4 данной главы.
В результате применения разработанного SPGF-метода нам впервые удалось описать колебательно – вращательную структуру (колебательно – вращательные переходы со значениями квантовых чисел Jмакс. =24 и Kaмакс. =19) всех известных на сегодняшний день полос поглощения одновременно для всех трех основных изотопических модификаций сероводорода, H2S, D2S и HDS (всего более 24000 линий) с точностью сопоставимой с погрешностями эксперимента. При этом число параметров модели было равно 549, что несопоставимо меньше, чем при попытке описать эти экспериментальные данные, например, в модели эффективных операторов (иные методы на современном этапе не позволяют этого сделать вообще).
Как отмечалось выше, основной целью исследования проведенного в параграфе 3.6 было выполнение расчетов потенциальной функции молекулы селеноводорода на основе экспериментальных данных о ее трех изотопических модификациях H2Se, D2Se и HDSe и сравнение результатов с результатами нашего же аналогичного исследования, выполненного в рамках довольно громоздкого MORBID метода. В качестве исходных данных нами были взяты те же экспериментальные данные что и при расчетах по MORBID модели. Полученные результаты позволяют сделать вывод, что развитый нами метод определения ВМПФ молекулы, дает результат не хуже чем MORBID метод, но требует несопоставимо меньше времени счета и компьютерной памяти (следует, однако, помнить, что в развитой в диссертации модификации метод применяется только к нормальным молекулам; в то же время MORBID метод применим и к молекулам с большой амплитудой колебаний).
Четвертая глава диссертации также посвящена исследованию спектров высокого разрешения молекул типа асимметричного волчка: H2O и H2CO. Основными причинами выделения этих молекул в отдельный раздел являются следующие. Молекула H2CO, будучи так же, как H2S и H2Sе, плоской молекулой типа асимметричного волчка, обладает важной особенностью по сравнению с двумя первыми. А именно, в ней возможны внеплоскостные (так называемые «booking») колебания, которые приводят хоть и к непринципиальным, но, тем не менее, изменениям в процессе реализации развитого нами метода определения ВМПФ. Как следствие, естественным является желание проверить работоспособность общего метода и на молекулах такого типа. Что касается молекулы H2O, то она, как известно, не является „нормальной“ из-за наличия довольно низкого потенциального барьера для деформационного колебания. Поэтому интересно было бы оценить границы применимости нашего метода и для молекул такого типа.
Первые два параграфа главы IV посвящены последовательно исследованию спектров высокого разрешения молекулы D2CO (как упоминалось ранее, наличие высокоточной экспериментальной информации о спектрах нескольких изотопических модификаций молекулы является необходимым условием корректного определения ее ВМПФ) и определению потенциальной функции молекулы формальдегида. Колебательно– вращательные спектры D2CO были зарегистрированы на Фурье-спектрометре Bruker IFS 120 HR в университете Оулу (Оулу, Финляндия) в диапазонах 650 – 1240 и 1780 - 2400 см-1 (в качестве иллюстрации спектр в диапазоне 1780 - 2400 см-1 приведен на рис.2), где расположены полиады из 3 и 9 сильно взаимодействующих друг с другом колебательных полос этой молекулы, соответственно.
В результате теоретического анализа экспериментального спектра было проинтерпретировано более 12000 линий указанных спектров, что позволило нам определить с высокой точностью (на уровне 0.0001 – 0.0002 см-1) как центры полос (что
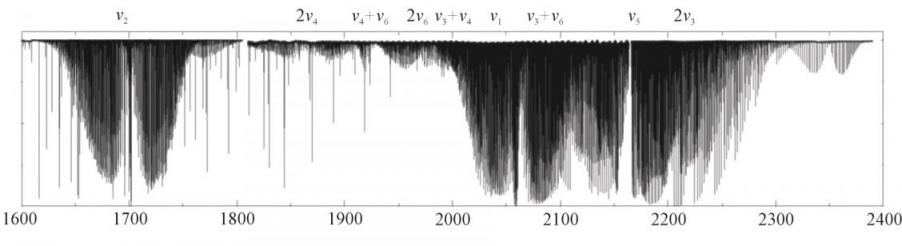
Рис. 2. Обзорный спектр молекулы D2CO.
является исходной информацией для задачи дальнейшего определения ВМПФ молекулы), так и колебательно – вращательные энергии всех колебательных состояний с максимальным значением квантового числа J = 48. Наличие сильных резонансных взаимодействий приводит к чрезвычайно запутанной картине энергетического спектра. Для ее описания требуется принять во внимание многочисленные резонансные эффекты, которые были учтены в эффективном операторе, описанном в разделе 4.1. Вместе с тем, наличие сильных резонансных взаимодействий позволило нам определить с высокой точностью несколько десятков колебательно – вращательных энергий, относящихся к запрещенной в поглощении по симметрии полосе ![]() , и центр этой полосы. Последнее обстоятельство должно быть особо отмечено, поскольку задача корректного определения вращательной структуры и центров запрещенных полос решается, как правило, с большими трудностями и требует специальных исследований (по этому поводу см., в частности, главу VI данной работы).
, и центр этой полосы. Последнее обстоятельство должно быть особо отмечено, поскольку задача корректного определения вращательной структуры и центров запрещенных полос решается, как правило, с большими трудностями и требует специальных исследований (по этому поводу см., в частности, главу VI данной работы).
Как показывает проведенный в разделе 4.2 анализ формул, лежащих в основе развитого метода определения ВМПФ, этот метод является весьма универсальным. Несмотря на необходимость введения для молекулы формальдегида так называемого «booking» внеплоскостного колебания, сильно отличающегося по своим характеристикам от рассмотренных в предыдущих главах валентных и деформационных колебаний, идеология метода остается неизменной. Определенные коррективы, которые обсуждаются в 4.2, следует сделать лишь в алгоритме расчета констант форм колебаний ![]() и коэффициентов
и коэффициентов ![]() ,
, ![]() в разложениях (1) и (2), а также определить зависимость «booking» колебания
в разложениях (1) и (2), а также определить зависимость «booking» колебания ![]() от колебательных координат
от колебательных координат ![]() :
:
![]() (12)
(12)
Три индекса в обозначении колебания ![]() появляются как следствие того, что он определяется как функция угла между связью C – O и плоскостью H – C – H.
появляются как следствие того, что он определяется как функция угла между связью C – O и плоскостью H – C – H.
Модифицированный на этой основе метод использовался для определения ВМПФ формальдегида. В качестве исходных данных были взяты центры 12-ти полос молекулы D2CO из раздела 4.1 и 240 экспериментальных (определены из спектров флюоресценции) центра полос молекулы H2CO. Как следствие решения задачи, мы определили 39 параметров модели, которые воспроизводят исходные центры полос со среднеквадратичным отклонением 0.97 см-1. Как и в предыдущих разделах работы, это может считаться вполне удовлетворительным результатом.
Следующие три параграфа главы IV посвящены исследованию тонкой структуры спектров дейтерированного и дважды дейтерированного водяного пара и оценкам параметров потенциальной функции молекулы H2O на основе экспериментальной информации о ее не сильно возбужденных по деформационному колебанию состояний. В разделах 4.4 и 4.5 приведены результаты теоретического анализа зарегистрированных впервые с высоким разрешением в широком спектральном диапазоне 3200 – 12900 см-1 спектров поглощения молекул HDO и D2O. До недавнего времени как интерпретация, так и, в особенности, дальнейший теоретический анализ в рамках модели эффективных операторов полученной из интерпретации спектров информации представляли большой интерес как с чисто академической точки зрения, так и в связи с практическими потребностями многочисленных прикладных задач. Причинами такого положения дел служили, в основном, два момента: В силу сложности и полного отсутствия какой бы то ни было регулярности, для выполнения корректной интерпретации спектров высокого разрешения водяного пара необходимо иметь хорошие предсказания положений линий. До недавнего времени единственной возможностью для такого рода предсказаний являлись результаты решения обратной спектроскопической задачи в рамках модели эффективных операторов. В свою очередь, наличие в спектрах молекулы H2O и ее дейтерированных модификаций многочисленных особенностей, неопределенностей и разнообразных резонансных ситуаций приводило к необходимости постоянного усовершенствования теоретической модели, положенной в основу метода эффективных операторов. В последние годы в связи с появлением работ Швенке, Теннисона и других, в которых энергетическая колебательно - вращательная структура всех изотопических разновидностей водяного пара была рассчитана с высокой точностью до значений более чем 25000 см-1 на основе информации о потенциальной функции молекулы H2O, использование эффективных гамильтонианов применительно к молекуле водяного пара, в значительной, степени потеряло смысл. Как следствие, и проблема интерпретации спектров водяного пара становится в настоящее время рутинной задачей. В то же время, на момент нашего исследования спектров D2O и HDO количественные результаты работ Швенке и Теннисона не были широко известны, поэтому полученные результаты представляли несомненный интерес.
В параграфе 4.3 в рамках «расширенной модели локальных мод» выполнено исследование эффекта изотопозамещения ![]() для трехатомных плоских молекул с произвольной величиной угла между связями. Получен ряд ранее неизвестных нетривиальных соотношений между различными спектроскопическими параметрами материнской и замещенной молекул. В качестве иллюстрации, одно из полученных соотношений приведено ниже:
для трехатомных плоских молекул с произвольной величиной угла между связями. Получен ряд ранее неизвестных нетривиальных соотношений между различными спектроскопическими параметрами материнской и замещенной молекул. В качестве иллюстрации, одно из полученных соотношений приведено ниже:
![]() , (13)
, (13)
где![]() - известная функция параметров основной изотопной модификации.
- известная функция параметров основной изотопной модификации.
Сложность спектров как молекулы D2O, так и HDO, резко возрастает при возрастании колебательных квантовых чисел рассматриваемых состояний. Поэтому для корректного описания положений колебательно – вращательных линий в высоковозбужденных состояниях нами были проведены специальные исследования и в теоретическую модель молекулы введены отличные от традиционных резонансов Ферми, Дарлинга – Деннисона и Кориолиса резонансные взаимодействия между состояниями ![]() и
и ![]() ,
, ![]() и
и ![]() и другие.
и другие.
Как и в случае сероводорода, экспериментальные спектры молекул D2O и HDO были впервые зарегистрированы с высоким разрешением на Фурье-спектрометре Bruker IFS 120 HR в университете наук и технологий Китая (Хефей, КНР). В общей сложности в разделах 4.4 – 4.5 диссертации приведены результаты интерпретации более чем 8000 колебательно – вращательных переходов и решения соответствующих обратных спектроскопических задач для ранее не исследовавшихся с высоким разрешением 21 полос молекулы D2O (полосы, входящие в состав полиад ![]() 2, 3,
2, 3, ![]() 4, 5) и 16 полос молекулы HDO (верхние колебательные состояния (101), (021), (210), (050), (130), (300), (111), (021), (060), (041), (201), (121), (070), (310), (150) и (230)).
4, 5) и 16 полос молекулы HDO (верхние колебательные состояния (101), (021), (210), (050), (130), (300), (111), (021), (060), (041), (201), (121), (070), (310), (150) и (230)).
Как отмечалось выше, в настоящее время потенциальная функция молекулы воды известна с настолько высокой точностью, что попытка даже ее воспроизведения на основе развитого в данной работе метода вряд ли будет успешной. Поэтому в разделе 4.6 мы, не конкурируя с результатами Швенке и Теннисона, поставили задачу проверить работоспособность разработанного нами метода на примере молекулы водяного пара с использованием, в качестве исходной, экспериментальной информации из двух предыдущих параграфов диссертации. Не останавливаясь на деталях, отметим лишь, что результаты проведенных расчетов показали, что даже для таких молекул как водяной пар, содержащих деформационные колебания большой амплитуды, разработанный нами метод дает приемлемые результаты вплоть до значений квантового числа деформационного колебания v, равного 6 – 7.
В пятой главе диссертации рассмотрен более сложный класс молекул, а именно, четырехатомные молекулы аксиальной симметрии типа XY3. Наличие более высокой симметрии по сравнению с молекулами, рассмотренными в предыдущих главах, приводит к ряду последствий, связанных с решением основной проблемы данной диссертационной работы. Одним из следствий является усложнение теоретического анализа экспериментальных спектров и, как следствие, экспериментальной основы реализации развитого нами метода определения ВМПФ. Вместе с тем, наличие в молекуле дважды вырожденных колебаний приводит к необходимости определенной, хотя и непринципиальной, модификации и самого метода.
Предметом исследования в данной главе являются молекулы (и их дейтеропроизвод-ные модификации) PH3, и AsH3. В разделах 5.1 – 5.2 и 5.4 – 5.6 рассматриваются вопросы, связанные с выполненными нами исследованиями спектров высокого разрешения молекул PH3, PH2D, PHD2 и PD3. Следует заметить, что спектры низкосимметричных изотопических модификаций обеих молекул ранее практически не исследовались. Поэтому понятным является желание компенсировать отсутствие какой бы то ни было начальной информации возможностью теоретического предсказания тех или иных величин, которые бы упростили процедуру интерпретации спектров. Для решения данной проблемы нами в рамках развитого ранее «расширенного метода локальных мод» был определен целый ряд нетривиальных изотопических соотношений, связывающих спектроскопические параметры «материнской» XH3 и низко-симметричных XH2D, и XHD2 модификаций. Эти вопросы рассматриваются в разделе 5.3.
Аналогичные исследования спектров молекул AsD3, AsH2D и AsHD2 представлены в разделе 5.7. В 5.8 рассмотрен разработанный нами и реализованный на примере молекулы фосфина оригинальный метод определения равновесных параметров молекулы на основе анализа свойств шпуров матриц эффективных гамильтонианов молекулы и/или ее дейтеропроизводных модификаций.
В последнем параграфе 5.9 обсуждаются особенности развитого в главе II метода применительно к аксиально-симметричным молекулам XY3, и метод применен к задачам определения ВМПФ рассматриваемых молекул.
Исследование колебательно – вращательного спектра молекулы PH3 в нашей работе было выполнено для спектрального диапазона 2000 – 4000 см-1, где расположены колебательно – вращательные полосы двух полиад: (а). полосы ![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() и ряд существенно более слабых полос поглощения; (б). полиада, состоящая из 18 полос, наиболее интенсивными из которых являются
и ряд существенно более слабых полос поглощения; (б). полиада, состоящая из 18 полос, наиболее интенсивными из которых являются ![]() ,
, ![]() ,
, ![]() и
и ![]() . Как и в ряде предыдущих случаев, спектр был зарегистрирован на Фурье-спектрометре Bruker IFS 120 HR в университете наук и технологий Китая.
. Как и в ряде предыдущих случаев, спектр был зарегистрирован на Фурье-спектрометре Bruker IFS 120 HR в университете наук и технологий Китая.
Поскольку молекула PH3 обладает достаточно высокой симметрией, то для анализа ее колебательно – вращательных спектров наиболее разумно и полезно использовать эффективный гамильтониан в симметризованной форме, который может быть построен на основании результатов и теорем теории неприводимых тензорных систем:
![]() . (13)
. (13)
Здесь суммирование ведется по всем резонирующим колебательным состояниям, ![]() – колебательные функции симметрии. Операторы
– колебательные функции симметрии. Операторы ![]() диагональных блоков описывают вращательные структуры соответствующих колебательных состояний. Операторы
диагональных блоков описывают вращательные структуры соответствующих колебательных состояний. Операторы ![]() ( ) недиагональных блоков связаны с различного рода резонансными взаимодействиями. Конкретный вид входящих в (13) операторов
( ) недиагональных блоков связаны с различного рода резонансными взаимодействиями. Конкретный вид входящих в (13) операторов ![]() довольно громоздок, поэтому здесь мы их не приводим. Важно, что используемый гамильтониан позволяет учесть все без исключения эффекты и взаимодействия, которые могут возникнуть в колебательно – вращательных спектрах аксиально – симметричных молекул.
довольно громоздок, поэтому здесь мы их не приводим. Важно, что используемый гамильтониан позволяет учесть все без исключения эффекты и взаимодействия, которые могут возникнуть в колебательно – вращательных спектрах аксиально – симметричных молекул.
Не останавливаясь подробно на деталях теоретического анализа спектра, в результате которого вращательная структура всех вышеупомянутых полос была проинтерпретирована вплоть до значений квантового числа J = 15, отметим лишь несколько, на наш взгляд, наиболее важных результатов. Во-первых, в плане исследования основной задачи данной диссертации, в результате теоретического анализа зарегистрированных спектров нам удалось впервые определить с высокой точностью центры полос ![]() ,
,![]() ,
, ![]() ,
, ![]() ,
, ![]() ,
, ![]() и уточнить центры полос
и уточнить центры полос ![]() и
и ![]() . Во-вторых, как результат корректного анализа спектра на основе использования симметризованного гамильтониана в форме (13), нам удалось обнаружить и проинтерпретировать линии запрещенной по симметрии полосы
. Во-вторых, как результат корректного анализа спектра на основе использования симметризованного гамильтониана в форме (13), нам удалось обнаружить и проинтерпретировать линии запрещенной по симметрии полосы ![]() . В третьих, впервые в практике спектроскопии были обнаружены и описаны а1/а2 расщепления для состояний (J K а1) /(J K а2) не только для значений
. В третьих, впервые в практике спектроскопии были обнаружены и описаны а1/а2 расщепления для состояний (J K а1) /(J K а2) не только для значений ![]() , но для K = 5, 6,…, вплоть до значений K = 10. Последние два обстоятельства являются следствием наличия в молекуле очень сильных резонансных взаимодействий, приводящих к многочисленным аномалиям в спектрах. Иллюстрация одной из многих таких аномалий, выявленных нами в результате теоретического анализа экспериментальных данных, приведена на рис. 3.
, но для K = 5, 6,…, вплоть до значений K = 10. Последние два обстоятельства являются следствием наличия в молекуле очень сильных резонансных взаимодействий, приводящих к многочисленным аномалиям в спектрах. Иллюстрация одной из многих таких аномалий, выявленных нами в результате теоретического анализа экспериментальных данных, приведена на рис. 3.
Поскольку молекула фосфина является самой легкой из пирамидальных аксиально-симметричных нормальных молекул, возможность корректного применения развитого нами в главе II мето-
Рис. 3. Иллюстрация аномального поведения а1/а2 расщеплений в зависимости от величины квантого числа J для состояний [J K=1 а1/а2](0002, E) (кривая I). Для сравнения нормальная зависимость а1/а2 расщеплений иллюстрируется кривой II.
да определения ВМПФ к этой молекуле будет означать возможность его корректного применения и к любой другой более тяжелой молекуле подобного типа. Вместе с тем, как уже отмечалось выше, для получения максимально правдоподобной ВМПФ необходимо иметь как можно больше экспериментальной информации о как можно большем числе изотопических модификаций. По этой причине нами были выполнены обширные исследования спектров высокого разрешения не только молекулы PH3, но и всех ее наиболее значимых изотопомеров.
Исследование возбужденных состояний молекулы в принципе невозможно без корректной информации о вращательной структуре основного колебательного состояния. В то же время, на момент начала нашего исследования низко-симметричных модификаций фосфина корректная информация о параметрах их основных колебательных состояний отсутствовала. Поэтому исследование спектров молекул PH2D и PHD2 было начато с определения корректных параметров их основных колебательных состояний. Для этого был специально зарегистрирован спектр высокого разрешения в длинноволновой области от 20 до 320 см-1. Поскольку анализ спектра традиционными методами оказался невозможным, нами был разработан специальный подход, известный в настоящее время как «метод двух пар переходов». В основу его был положен комбинационный принцип Рица. Будучи реализован в виде программ для ЭВМ, разработанный метод позволил нам с высокой точностью определить чисто вращательный спектр обеих низко-симметричных модификаций фосфина. Сказанное выше является предметом раздела 5.2.
В параграфе 5.3 рассматриваются вопросы дальнейшего развития теории изотопозамещения для молекул, удовлетворяющих модели «расширенного метода локальных мод», к которым и относятся фосфин и арсин. Не останавливаясь подробно на преобразованиях, описанных в параграфе 5.3, отметим, что результатом проведенных исследований явился целый ряд ранее неизвестных соотношений, позволяющих выполнять расчеты предсказательного характера спектроскопических параметров молекул XH2D и XHD2 по известным экспериментальным значениям параметров значительно лучше изученных «материнских» модификаций XH3. При этом, как показало сравнение с реальными значениями соответствующих параметров, полученных позже из анализа экспериментальных данных, точность предсказания для различных параметров колеблется в пределах 15-30 процентов. В условиях полного отсутствия какой бы то ни было информации такая точность предсказания представляется вполне удовлетворительной. В качестве иллюстрации ниже приведено одно из полученных соотношений:
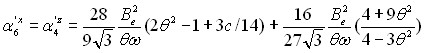 (14)
(14)
Здесь ![]() и
и ![]() - равновесная вращательная постоянная и деформационная гармоническая частота молекулы XH3;
- равновесная вращательная постоянная и деформационная гармоническая частота молекулы XH3; ![]() и
и ![]() - известные функции параметров основной модификации. Полученные в данном параграфе результаты использовались затем в качестве предварительных оценок параметров при анализе реальных спектров.
- известные функции параметров основной модификации. Полученные в данном параграфе результаты использовались затем в качестве предварительных оценок параметров при анализе реальных спектров.
Разделы 5.4 и 5.5 посвящены обширным исследованиям спектров высокого разрешения молекул PH2D и PHD2, зарегистрированных в университете Вупперталя в диапазоне от 600 до 5000 см-1. Предметом исследования явились 23 колебательно–вращательные полосы, соответствующие всем однократным и практически всем двукратным возбуждениям колебательных квантовых чисел в молекуле PH2D и 6 полос (![]() ,
,![]() ,
,![]() ,
,![]() ,
,![]() и
и![]() ) молекулы PHD2. Последняя была зарегистрирована на высокочувстви-тельном лазерном фотоакустическом спектрометре университета г. Лилля (Франция). Совместное использование результатов, приведенных в разделах 5.2 и 5.3, и модифицированного для описания молекул типа XH2D и XHD2 эффективного гамильтониана позволило описать все зарегистрированные спектры с точностью близкой к экспериментальной, то есть на уровне 0.0001 – 0.0003 см-1 для Фурье-спектров и на уровне 0.002 – 0.008 см-1 для полосы
) молекулы PHD2. Последняя была зарегистрирована на высокочувстви-тельном лазерном фотоакустическом спектрометре университета г. Лилля (Франция). Совместное использование результатов, приведенных в разделах 5.2 и 5.3, и модифицированного для описания молекул типа XH2D и XHD2 эффективного гамильтониана позволило описать все зарегистрированные спектры с точностью близкой к экспериментальной, то есть на уровне 0.0001 – 0.0003 см-1 для Фурье-спектров и на уровне 0.002 – 0.008 см-1 для полосы ![]() . Анализ полученных из решения обратных спектроскопических задач результатов позволил сделать вывод, что принадлежность основной изотопической модификации XH3 к объектам, удовлетворяющим модели локальных мод, приводит для низкосимметричных модификаций к возможности рассмотрения двух фрагментов XH2/XD2 и XD/XH как достаточно независимых друг от друга квазимолекул. При этом фрагмент XH2/XD2 показывает свойства, характерные для трехатомных молекул, удовлетворяющих условиям модели локальных мод, а фрагмент XD/XH – свойства характерные для двухатомных молекул. Этот вывод является важным для будущих исследований высоковозбужденных колебательных состояний молекул XH2D и XHD2. Выполненный здесь же теоретический анализ вращательной структуры полосы
. Анализ полученных из решения обратных спектроскопических задач результатов позволил сделать вывод, что принадлежность основной изотопической модификации XH3 к объектам, удовлетворяющим модели локальных мод, приводит для низкосимметричных модификаций к возможности рассмотрения двух фрагментов XH2/XD2 и XD/XH как достаточно независимых друг от друга квазимолекул. При этом фрагмент XH2/XD2 показывает свойства, характерные для трехатомных молекул, удовлетворяющих условиям модели локальных мод, а фрагмент XD/XH – свойства характерные для двухатомных молекул. Этот вывод является важным для будущих исследований высоковозбужденных колебательных состояний молекул XH2D и XHD2. Выполненный здесь же теоретический анализ вращательной структуры полосы ![]() молекулы XHD2 полностью подтвердил сделанный вывод.
молекулы XHD2 полностью подтвердил сделанный вывод.
Теоретическому исследованию первой обертонной ![]() и комбинационной
и комбинационной ![]() полос полностью дейтерированной модификации PD3 (спектр также зарегистрирован в Вуппертале, Германия) посвящен параграф 5.6. Ранее инфракрасные спектры молекулы PD3 регистрировались и изучались только в области фундаментальных полос 1, 2, 3 и 4. Что касается обертонных и комбинационных полос, то они исследованы в нашей работе впервые. Заметим, что полосы
полос полностью дейтерированной модификации PD3 (спектр также зарегистрирован в Вуппертале, Германия) посвящен параграф 5.6. Ранее инфракрасные спектры молекулы PD3 регистрировались и изучались только в области фундаментальных полос 1, 2, 3 и 4. Что касается обертонных и комбинационных полос, то они исследованы в нашей работе впервые. Заметим, что полосы ![]() и
и ![]() имеют разную симметрию и разность между их центрами всего 2.8 см-1. Это приводит к гигантским резонансным взаимодействиям Кориолисова типа, которые должны быть учтены в модели молекулы, уже при значениях квантового числа J = 3 - 4. Как и в предыдущих разделах работы, теоретический анализ в рамках той же модели гамильтониана, что и для молекулы PH3, позволил описать зарегистрированный спектр в точностью на уровне погрешностей эксперимента. В качестве интересного факта, выявленного в процессе анализа спектра, следует отметить, что полученные в результате спектроскопические параметры однозначно указывают на принадлежность молекулы PD3 к молекулам, удовлетворяющим модели локальных мод, что априори не являлось очевидным до выполнения нашего исследования. Последнее, так же как и выше для низко-симметричных изотопических модификаций, позволяет существенно облегчить анализ спектров молекулы PD3 в высоковозбужденных состояниях.
имеют разную симметрию и разность между их центрами всего 2.8 см-1. Это приводит к гигантским резонансным взаимодействиям Кориолисова типа, которые должны быть учтены в модели молекулы, уже при значениях квантового числа J = 3 - 4. Как и в предыдущих разделах работы, теоретический анализ в рамках той же модели гамильтониана, что и для молекулы PH3, позволил описать зарегистрированный спектр в точностью на уровне погрешностей эксперимента. В качестве интересного факта, выявленного в процессе анализа спектра, следует отметить, что полученные в результате спектроскопические параметры однозначно указывают на принадлежность молекулы PD3 к молекулам, удовлетворяющим модели локальных мод, что априори не являлось очевидным до выполнения нашего исследования. Последнее, так же как и выше для низко-симметричных изотопических модификаций, позволяет существенно облегчить анализ спектров молекулы PD3 в высоковозбужденных состояниях.
В параграфе 5.7 выполнено аналогичное рассмотренному выше исследование ряда впервые зарегистрированных в университете Вупперталя спектров высокого разрешения молекул AsD3, AsHD2 и AsH2D. Не останавливаясь здесь, ввиду ограниченности объема, на деталях, заметим лишь, что как молекулы – основные изотопические модификации фосфина и арсина, так и их различные изотопические производные обладают весьма схожими спектроскопическими свойствами. Поэтому выводы сделанные в разделе 5.7 при анализе спектров изотопомеров арсина полностью соответствуют выводам сделанным ранее при анализе спектров фосфина.
Поскольку структурные параметры, наряду с параметрами ВМПФ, являются фундаментальными характеристиками молекул, как можно более точному определению их в спектроскопии всегда уделялось особое внимание. В разделе 5.8 обсуждается предложенный нами новый метод определения равновесных параметров многоатомной молекулы. Показано, что использование свойств шпуров матриц позволяет выразить значения равновесных углов и длин связей в виде функций непосредственно экспериментальных значений колебательно–вращательных энергий нескольких изотопических модификаций молекулы. Проанализировано влияние различного рода причин, приводящих к возможным погрешностям метода. В качестве иллюстрации работоспособности, разработанный общий подход применен к задаче определения равновесной структуры молекулы PH3 на основе полученных в предыдущих разделах результатов для молекул PH2D и PHD2.
В параграфе 5.9 анализируются особенности применения представленного в главе II метода определения ВМПФ к пирамидальным аксиально-симметричным молекулам типа XH3. Показано, что изменения не носят принципиального характера и сводятся лишь к несущественным изменениям алгоритма расчетов, что может рассматриваться как еще одно подтверждение универсальности подхода. Модифицированный алгоритм используется затем для численного расчета ВМПФ молекул фосфина и арсина. При этом в качестве исходной информации для молекулы арсина были использованы как наши собственные результаты теоретического анализа экспериментальных спектров, так и литературные данные. Что касается молекулы фосфина, то, имея в виду значительное количество новой информации по спектрам фосфина, полученной в нашей работе, в качестве исходных данных были использованы только наши результаты. Литературные же данные по спектрам молекулы PH3 рассматривались в качестве теста предсказательной возможности полученных нами результатов. Как для молекулы фосфина, так и для молекулы арсина полученные в результате параметры потенциальных функций воспроизводят исходные экспериментальные данные со средней точностью лучше чем 1 см-1. Сравнение полученных результатов с известными в литературе данными показывает вполне удовлетворительное их согласие, хотя при этом наши результаты лучше согласуются с экспериментальными данными.
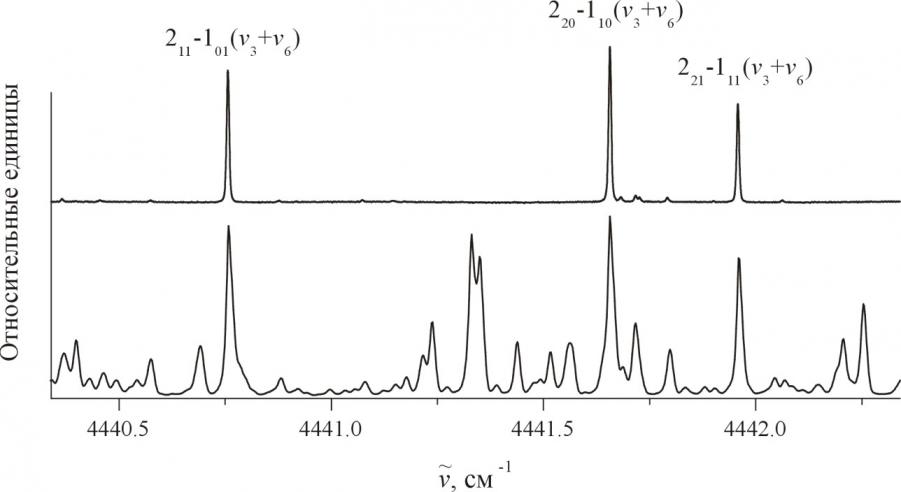
Заключительная глава 6 диссертации посвящена рассмотрению еще более сложного со спектроскопической точки зрения объекту – молекуле метана. Молекула метана является уже пятиатомной молекулой, и до настоящего времени задача о корректном определении ее потенциальной функции не была решена. Причины заключаются в том, что из-за наличия в молекуле достаточно большого числа атомов (а). усложняется процедура построения ЕКЕ операторов и (б). резко возрастают размеры матрицы гамильтониана. Еще одним немаловажным ограничением для успешного решения задачи определения ВМПФ метана является ограниченное количество данных, которые можно было бы использовать как исходную экспериментальную информацию. Поэтому первым шагом нашего исследования была регистрация спектров высокого разрешения молекул CH3D, CH2D2 и CHD3 в широком спектральном диапазоне 2700 – 6300 см-1. Спектры были зарегистрированы на Фурье-спектрометре Bruker IFS 120 HR (ZP 2001) в федеральном институте высоких технологий Швейцарии (Цюрих, Швейцария) при температуре 78 K. Последнее обстоятельство является важным, поскольку сильно упрощает задачу интерпретации спектра и поиска новых полос поглощения (напомним, что для решения нашей центральной задачи основной целью анализа спектров является не регистрация как можно большего числа линий, а корректное определение как можно большего числа центров полос). В качестве иллюстрации сказанного на рис. 4 приведен небольшой фрагмент зарегистрированного нами спектра молекулы CH2D2 при температуре 78 K и, для сравнения, тот же участок спектра, зарегистрированный при комнатной температуре.
В результате исследования зарегистрированных спектров нами было обнаружено и описано почти 200 новых полос поглощения для
Рис.4. Фрагмент спектра высокого разрешения молекулы CH2D2 в районе полосы v3+v6: вверху спекр, зарегистрированный на спектрометре Bruker IFS 120 HR (ZP 2001), 78К; внизу – BOMEM, комнатная температура.
молекул CH2D2, CH3D и CHD3. Будучи добавлены к 38 известным до нашего исследования центрам полос этих молекул, они послужили хорошей основой для решения задачи определения ВМПФ метана.
Следует заметить, что молекула метана обладает существенно более высокой симметрией, нежели рассмотренные ранее молекулы. Поэтому описанный в главе два общий метод был модифицирован с учетом этого условия, а именно, формулы и выводы, составляющие основу метода были переформулированы в терминах теории неприводимых тензорных систем. Показано, что, за исключением этого обстоятельства, вся схема метода определения ВМПФ и в этом случае претерпевает лишь малые изменения, и может быть использована для решения проблемы. Единственное принципиальное отличие рассматриваемой молекулы от исследованных ранее молекул заключается в наличии у молекулы метана так называемого эффекта «redundancy», который чрезвычайно усложняет процедуру использования естественных координат. Поэтому, применительно к молекуле метана, мы использовали в расчетах координаты симметрии ![]() и получили параметры потенциальной функции, восстанавливающие исходные экспериментальные данные с точностью порядка 0.8 см-1. Следует заметить, что любой из известных в настоящее время аналогичных результатов не позволяет этого сделать одновременно для трех изотопических модификаций.
и получили параметры потенциальной функции, восстанавливающие исходные экспериментальные данные с точностью порядка 0.8 см-1. Следует заметить, что любой из известных в настоящее время аналогичных результатов не позволяет этого сделать одновременно для трех изотопических модификаций.
Наличие в молекуле метана девяти колебательных степеней свободы приводит к чрезвычайно запутанной картине даже колебательных спектров ее дейтерозамещенных модификаций, не говоря уже о колебательно-вращательной структуре. Как следствие, для выявления особенностей энергетических структур трех рассматриваемых в диссертации модификаций метана, решению основной задачи определения ВМПФ метана в главе VI предшествует теоретический анализ полученных из экспериментальных данных как колебательно – вращательных, так и чисто колебательных энергий в рамках более простых моделей, в том числе в адаптированной к исследованию больших совокупностей колебательных полос высоко-симметричных молекул модели эффективных операторов.
В качестве практического приложения основного результата шестой главы, полученная нами потенциальная функция использовалась в ряде задач, которые ранее невозможно было решить ввиду различного рода причин и которые оказалось возможным устранить при наличии корректной ВМПФ метана. В диссертации, в частности, рассматриваются две из таких задач. Первая - совместный анализ 22-х сильно взаимодействующих полос молекулы CH2D2, расположенных в районе 2000 – 3100 см-1. В этом случае знание ВМПФ позволило теоретически предсказать все наиболее важные параметры данной совокупности полос (центры, вращательные и наиболее важные из резонансных параметров) и как следствие решить обратную спектроскопическую задачу, которая в течение долгого времени была не решена в полном объеме из-за наличия многочисленных и разнообразных резонансных взаимодействий и корреляций между параметрами. Второе приложение полученной в диссертации ВМПФ метана – существенно более сложная задача описания колебательно–вращательной структуры 89-ти образующих «колебательный квазиконтинуум» полос молекулы CH2D2 в районе 3100 – 4000 см-1. Основным моментом, позволившим так же как и выше, решить задачу в данном случае, явилась возможность предсказания всех основных параметров модели на основе информации о найденной нами потенциальной функции.
В заключении сформулированы основные выводы и результаты проведенных исследований:
- Развит оригинальный метод определения внутримолекулярной потенциальной функции многоатомной нормальной молекулы на основе экспериментальной информации о колебательно – вращательных спектрах ее различных изотопических модификаций. Наряду с эффективностью разработанный метод характеризуется значительной простотой при реализации по сравнению с известными на настоящий момент аналогичными подходами и применим для молекул с произвольным числом атомов и произвольной симметрии.
- Разработана и реализована в виде алгоритмов и программ на языках MAPLE и FORTRAN расчетная схема решения систем нелинейных уравнений (вплоть до нескольких сотен), позволяющая определять константы форм колебаний произвольных многоатомных молекул.
- На основе использования трансформационных свойств различных колебательных координат, использующихся в колебательно – вращательной спектроскопии многоатомных молекул, разработан метод построения точного оператора для «кинетической» части гамильтониана молекулы, который, не уступая в корректности известным в теоретической спектроскопии ЕКЕ («exact kinetic energy») операторам, в отличие от них (а). является универсальным, то есть может использоваться применительно к молекулам с любым числом атомов и симметрии; (б). существенно проще ЕКЕ-операторов при выполнении практических расчетов; (с). применим к любым изотопическим модификациям рассматриваемой молекулы.
- Проведено исследование условий возможности редукции матрицы гамильтониана произвольной многоатомной молекулы и выработаны количественные критерии проведения той или иной формы редукции. На этой основе разработан и практически реализован способ, позволяющий с разумными компьютерными мощностями и временными затратами реализовывать итерационные процедуры для задач, требующих многократного построения и дальнейшей диагонализации матриц большой размерности.
- Предложен и практически реализован «метод двух пар переходов», позволяющий определять вращательную структуры основного колебательного состояния для широкого класса молекул, обладающих симметрий CS.
- Получила дальнейшее развитие теория изотопозамещения в многоатомных молекулах: для молекул, удовлетворяющих условиям «расширенной модели локальных мод», получены изотопические соотношения, позволяющие предсказывать значения различных спектроскопических параметров (гармонических частот, параметров ангармоничности, вращательных постоянных и колебательно - вращательных коэффициентов, различного рода резонансных параметров) низко-симметричных изтопических модификаций по экспериментальным значениям спектроскопических параметров, как правило, существенно лучше изученных «материнских» модификаций, обладающих большей симметрией.
- Показано, что в возбужденных колебательных состояниях фрагменты XH2/ XD2 и XD/XH низко-симметричных изотопических модификаций аксиально-симметричных молекул XH3 можно рассматривать как независимые. Как следствие, спектроскопические свойства молекул XH2D и XHD2 в высоковозбужденных состояниях подобны свойствам удовлетворяющих модели локальных мод трехатомных молекул XH2/XD2, с одной стороны, и двухатомных молекул типа XD/XH, с другой стороны.
- Впервые при теоретическом анализе спектров молекулы PH3 были обнаружены и описаны а1/а2 расщепления для состояний (J K а1) /(J K а2) не только для значений квантового числа
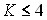 , но для K = 5, 6,…, вплоть до значений K = 10.
, но для K = 5, 6,…, вплоть до значений K = 10. - Для молекул близких к «сплющенному» симметричному волчку показана корректность и эффективность использования эффективного гамильтониана в А-редукции и IIIl-представлении для описания их колебательно-вращательной структуры.
- Получил дальнейшее развитие метод «глобального фиттинга». Выполнена его модернизация применительно к широкому классу молекул, обладающих симметрией Cs. На основе усовершенствованного метода выполнен одновремен-ный колебательно-вращательный анализ всех известных на сегодняшний день колебательно-вращательных спектров молекулы HDS.
- Развит и на примере трехатомных молекул H2S/HDS/D2S практически реализован SPGF (Spectroscopic Potential – Global Fit) метод, являющийся дальнейшим развитием программы по разработке эффективных и простых методов определения потенциальных многомерных поверхностей многоатомных молекул и представляющий собой компиляцию ранее разработанных метода «глобального фиттинга» и метода определения ВПМФ, описанного в главе II.
- На основе анализа впервые зарегистрированных в широком спектральном диапазоне 2800 – 6500 см-1 охлажденных спектров высокого разрешения молекул CH2D2, CH3D, и CHD 3 получена информация о почти 200-х новых колебательно-вращательных полосах этих молекул, которая позволила корректно решить задачу определения потенциальной функции метана.
- На этой основе впервые выполнен совместный анализ большого числа колебательно-вращательных полос молекулы CH2D2.
- В рамках развитого общего метода определены потенциальные функции молекул различной симметрии, содержащих различное количество атомов: H2S, H2Se, PH3, AsH3, H2CO и CH4, позволяющие одновременно описывать колебательные спектры всех изотопических модификаций соответствующих молекул.
- Впервые выполнен теоретический анализ в общей сложности более чем 400 впервые зарегистрированных с высоким разрешением колебательно – вращательных полос (более 80000 линий поглощения, зарегистрированных с точностями в центах поглощения на уровне 1- 5 х 10-4 см-1 ) молекул H2S, D2S, HDS, H2Se, D2O, HDO, PH3, PH2D, PHD2, PD3, AsH2D, AsHD2, AsD3, D2CO, CH2D2, CH3D, CHD3
Таким образом, в диссертации разработана совокупность теоретических методов, направленных на решение одной из важнейших проблем современной колебательно – спектроскопии многоатомных молекул и позволяющих рассмотреть эту проблему с разных сторон, а именно, проблемы корректного определения внутримолекулярных многомерных потенциальных поверхностей (функций). Работоспособность и эффективность разработан-ных методов иллюстрируется многочисленными практическими приложениями к молекулам различного типа симметрии, содержащим разное число атомов.
Основные публикации по теме диссертации
- Ulenikov O. N., Wang Xiang-huai, Onopenko G.A., Bekhtereva E.S., He Sheng-gui, Hu Shui-ming, Hai Lin, Zhu Qing-shi. High Resolution Study of the First Hexad of D2O // J. Mol. Spectrosc. – 2000. – Vol. 200. P. 25–33.
- He Sheng-gui, Ulenikov O. N., Onopenko G.A., Bekhtereva E.S., Wang Xiang-huai, Hu Shui-ming, Hai Lin, Zhu Qing-shi. High Resolution Fourier Transform Spectrum of the D2O Molecule in the Region of the Second Triad of Interacting Vibrational States // J. Mol. Spectrosc. – 2000. – Vol. 200. P. 34–39.
- Hu Shui-ming, Ulenikov O. N., Onopenko G.A., Bekhtereva E.S., He Sheng-gui, Wang Xiang-huai,, Hai Lin, Zhu Qing-shi. High Resolution Study of Strongly Iiteracting Vibrational Bands of HDO in the Region of 7600 - 8100 cm-1 // J. Mol. Spectrosc. – 2000. – Vol. 203. P. 228–234.
- Ulenikov O. N., He Sheng-gui, Onopenko G.A., Bekhtereva E.S., Wang Xiang-huai, Hu Shui-ming, Hai Lin, Zhu Qing-shi. High Resolution Study of the (1+2 /2 + +3 = 3) Poliad of Strongly Interacting Vibrational Bands of D2O // J. Mol. Spectrosc. – 2000. – Vol. 204. P. 216–225.
- Zheng Jing-jing, Ulenikov O. N., Onopenko G.A., Bekhtereva E.S., He Sheng-gui, Wang Xiang-huai, Hu Shui-ming, Hai Lin, Zhu Qing-shi. High Resolution Vibration - Rotation Spectrum of the D2O Molecule in the Region Near the 21 + 2 + 3 Absorption Band // Molec. Phys. – 2001. – Vol. 99. P. 931–937.
- Ulenikov O. N., Hu Shui-ming, Bekhtereva E.S., Onopenko G.A., Wang Xiang-huai, He Sheng-gui, Zheng Jing-jing, Zhu Qing-shi. High Resolution Fourier Transform Spectrum of HDO in the Region of 6140 - 7040 cm-1 // J. Mol. Spectrosc. – 2001. – Vol. 208. P. 224–235.
- Ulenikov O. N., Onopenko G.A., Bekhtereva E.S., Sinitsin E.A., Brger H., Jerzembeck W. Isotopic Effects in the XH3 (C3v) Molecules: The Lowest Vibrational Bands of PH2D // J. Mol. Spectrosc. – 2001. – Vol. 208. P. 236–248.
- Zheng Jing-jing, Ulenikov O. N., Bekhtereva E. S., Ding Yun, He Sheng-gui, Hu Shui-ming, Wang Xiang-huai, Zhu Qing-shi. High Resolution Rotational Analysis of Deuterated Hypochlorous Acid: Ground State, (100) and (020) Vibrational Bands // J. Mol. Spectrosc. – 2001. – Vol. 209. P. 105–115.
- Ding Yun, Ulenikov O. N., Bekhtereva E. S., Zheng Jing-jirg, He Sheng-gui, Hu Shui-ming, Wang Xiang-huai, Zhu Qing-shi. High Resolution Rotational Analysis of the Lowest D - O Overtone Bands of Deuterated Hypochlorous Acid: 2v1 and 3v1 // J. Mol. Spectrosc. – 2001. - Vol. 209. - P. 233–241.
- Ulenikov O. N., Hu Shui-ming, Bekhtereva E. S., Onopenko G. A., Wang Xiang-huai, He Sheng-gui, Zheng Jing-jing, Zhu Qing-shi. High Resolution Fourier Transforg Spectrum of D2O in the Region Near 0.97 µm // J. Mol. Spectrosc. – 2001. - Vol. 210. - P. 18-27.
- Онопенко Г. А., Бехтерева Е.С., Синицын Е.А., Юрченко С. Н., Мельников В. В., Улеников О. Н. О некоторых проявлениях эффекта изотопозамещения в аксиально – симметричных молекулах: XH3 – XH2D // Оптика атмосферы и океана. – 2001. – Т. 14 – С. 134-136.
- Онопенко Г. А., Бехтерева Е.С., Синицын Е.А., Юрченко С. Н., Мельников В. В., Улеников О. Н. Об изотопическом эффекте в XH2 (C2v) молекулах с произвольной величиной равновесного угла
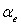 : XH2 - XHD // Оптика атмосферы и океана. – 2001. – Т. 14 – С. 212-214.
: XH2 - XHD // Оптика атмосферы и океана. – 2001. – Т. 14 – С. 212-214. - Бехтерева Е.С., Онопенко Г. А., Синицын Е.А., Улеников О. Н. Об определении равновесных структурных параметров PH3 // Оптика атмосферы и океана. – 2001. – Т. 14 – С. 215-217.
- Ulenikov O. N., Brger H., and Jerzembeck W., Onopenko G. A., Bekhtereva E. S., Petrunina O. L. On the Ground Vibrational States of the PH2D and PHD2 Molecules // J. Mol. Struct. – 2001. - Vol. 599. - P. 225-237.
- Hu Shui-ming, Ulenikov O. N., Bekhtereva E. S., Onopenko G. A., He Sheng-gui, Lin Hai, Cheng Ji-Xin, Zhu Qing-shi. High Resolution Fourier Transform Intra-Cavity Laser Absorption Spectroscopy of D2O in the Region of the 4v1+v3 Band // J. Mol. Spectrosc. – 2002. - Vol. 212. - P. 89-95.
- Ulenikov O. N., Bekhtereva E. S., Petrunina O. L., Brger H., Jerzembeck W. High Resolution Study of the Three Lowest Infrared Bands of PHD2 // J. Mol. Spectrosc. – 2002. - Vol. 214. - P. 1-10.
- Ulenikov O. N., Petrunina O. L., Bekhtereva E. S., Sinitsin E. A., Brger H., Jerzembeck W. High Resolution Infrared Study of PHD2: The P - H Stretching Bands v1 and 2v1 // J. Mol. Spectrosc. – 2002. - Vol. 215. - P.85-92.
- Ulenikov O. N., Bekhtereva E. S., Kozinskaia V. A., Zheng Jing-jing, He Sheng-gui, Hu Shui-ming, Zhu Qing-shi, Leroy C., Pluchart L. On the Study of Resonance Interactions and Splittings in the PH3 Molecule: v1, v3, v2+v4, and 2v4 Bands // J. Mol. Spectrosc. - 2002. - Vol. 215. - P. 295-308.
- Ulenikov O. N., Bekhtereva E. S., Onopenko G. A., Sinitsin E. A. On the Determination of the Equilibrium Structure of the PH3 Molecule // J. Mol. Spectrosc. – 2002. - Vol. 216. - P. 252-258.
- Ulenikov O. N., Bekhtereva E. S., Khabibulina O. L., Brger H., Jerzembeck W. Rotational Analysis of the v2 and 2v2 P - D Stretching Bands of PH2D // J. Mol. Spectrosc. – 2003. - Vol. 217. - P. 288-297.
- Ulenikov O. N., Bekhtereva E. S., Khabibulina O. L., Brger H., Jerzembeck W. High Resolution Study of the v1/v5 and 2v1/v1+v5 P - H Stretching Bands of PH2D // J. Mol. Spectrosc. – 2003. - Vol. 219. - P.13-29.
- Ulenikov O. N., Yuhnik Yu. B., Bekhtereva E. S., Tyabaeva N. E., H.Brger, Jerzembeck W., Fusina L. High Resolution Fourier Transform Spectrum of PD3 in the Region of the Stretching Overtone Bands 2v1 and v1+v3 // J. Mol. Spectrosc. – 2003. - Vol. 221. - P. 250-260.
- Ulenikov O. N., Bekhtereva E. S., Homiak T. D., Huet T. R., Herregodts F., Brger H., Jerzembeck W. High Resolution Study of the 6v1 P - H Stretching Band of the PHD2 Molecule // J. Mol. Spectrosc. – 2003. - Vol. 222. - P.153-158.
- Ulenikov O. N., Bekhtereva E. S., Kozinskaia V. A., Zheng Jing-jing, He Sheng-gui, Hu Shui-ming, Zhu Qing-shi, Leroy C., Pluchart L. High Resolution Spectrum of the v1+v4(E), v1+v4(E), v3+v4(E), v3+v4(A1), and v3+v4(A2) Bands of the PH3 Molecule: Assignment and Preliminary Analysis // JQSRT – 2004. - Vol. 83. - P. 599-618.
- Ulenikov O. N., Bekhtereva E. S., Grebneva S. V., Brger H., Jerzembeck W., Jerzembeck W., Leroy C. High Resolution Study of Some Doubly Excited Vibrational States of PH2D: The v1+v2, v2+v5, v2+v3, and v2+v6 Bands // J. Mol. Spectrosc. – 2004. - Vol. 226. - P.7-23.
- Ulenikov O. N., Liu A.-W., Bekhtereva E. S., Gromova O. V., Hao L.-Y., Hu S.-M. On the Study of High Resolution Rovibrational Spectrum of H2S in the Region of 7300 - 7900 cm-1 // J. Mol. Spectrosc. – 2004. - Vol. 226. - P.57-70.
- Ulenikov O. N., Bekhtereva E. S., Sanzharov N. A., Per Jensen. A Refined Potential Energy Function for the Electronic Ground State of H2Se // J. Mol. Spectrosc. – 2004. - Vol. 227. - P.1-12.
- Ulenikov O. N., Liu A.-W., Bekhtereva E. S., Grebneva S. V., Deng W.-P., Gromova O. V., Hu S.-M. High-Resolution Fourier Transform Spectrum of H2S in the Region of 8500 - 8900 cm-1 // J. Mol. Spectrosc. – 2004. - Vol. 228. - p. 110-119.
- Ulenikov O. N., Bekhtereva E. S., Grebneva S. V., Hollenstein H., Quack M. High Resolution Fourier Transform Spectrum of CH2D2 in the Region of 2350 - 2650 cm-1: The Bands v5+v7, 2v9, v3+v4, v3+v7, and v5+v9 // Phys. Chem. Chem. Phys. – 2005. - Vol.7(6). - P.1142-1150.
- Ulenikov O. N., Hu S.-M., Bekhtereva E. S., Zhu Q.-S. On the Study of High Resolution Rovibrational Spectrum of HDO in the Region of 8900 - 9500 cm-1: Some Remarks about "Effective Hamiltonians" Conception // J. Mol. Spectrosc. – 2005. - Vol. 231. - P.57-65.
- Lohilahti J., Ulenikov O. N., Bekhtereva E. S., Horneman V.-M., Alanko S. The Fundamental Bands v3/v4, and v6 of D213CO // J. Mol. Spectrosc. – 2005. - Vol. 231. - P. 108-116.
- Leroy C., Ulenikov O. N., Bekhtereva E. S., Onopenko G. A., Chudinova T. D. High-Resolution Study of the Six Lowest Doubly Excites Vibrational States of PH2D // J. Mol. Spectrosc. – 2005. - Vol. 234. - P.228-237.
- Ulenikov O. N., Liu A.-W., Bekhtereva E. S., Gromova O. V., Hao L.-Y., Hu S.-M. High-Resolution Fourier Transform Spectrum of H2S in the Region of the Second Hexade // J. Mol. Spectrosc. – 2005. - Vol. 234. - P.287-295.
- Юхник Ю.Б., Бехтерева Е.С. Спектр высокого разрешения AsD3 в районе полос поглощения 1 и 3 // Известия вузов. Физика. – 2005. – № 12 – С. 95. Деп. в ВИНИТИ 13.07.2005 г. № 1019-В 2005.
- Ulenikov O. N., Bekhtereva E. S., Bulavenkova A. S., Albert S., Bauerecker S., Hollenstein H., Quack M. High resolution spectroscopy and dynamics of the methane molecule: CH3D isotopomer // Abstr. 19th Colloquium on High Resolution Molecular Spectroscopy. – 2005. Salamanka, Spain. - P.N11.
- Ulenikov O.N., Yukhnik Yu.B., Bekhtereva E.S., Brger H. High resolution infrared study of the 1 and 3 bands, and the equilibrium structure of AsD3 // J. Mol. Struct. 2006. – Vol. 780-781. P. 115–123.
- Lohilahti J., Ulenikov O. N., Bekhtereva E. S., Alanko S., Anttila R. High Resolution Infrared Study of D2CO in the Region of 1780-2400 cm-1: Assignment and Preliminary Analysis // J. Mol. Struct. – 2006. - Vol. 780-781. - P.182-205.
- Ulenikov O. N., Bekhtereva E. S., Bulavenkova A. S., Leroy C., Brger H. High Resolution Study of AsHD2: Ground State and Three Lowest Bending Fundamental Bands, v3, v4, and v6 // J. Mol. Spectrosc. – 2006. - Vol.240. - P.102-111.
- Ulenikov O. N., Bekhtereva E. S., Grebneva S. V., Hollenstein H., Quack M. High Resolution Ro-Vibrational Analysis of Vibrational States of A2 Symmetry of the Dideuterated Methane CH2D2: States v5 and v7+v9 // Molec. Phys. – 2006. - Vol.104. - P.3371-3386.
- Liu A.-W., Ulenikov O. N., Onopenko G. A., Gromova O. V., Bekhtereva E. S., L. Wan, Hao L.-Y., Hu S.-M., Flaud J.-M. Global fit of the high resolution infrared spectrum of D2S // J. Mol. Spectrosc. – 2006. - Vol.240. - P.32-44.
- Ulenikov O.N., Liu A.-W., Bekhtereva E. S., Onopenko G. A., Gromova O. V., L. Wan, Hu S.-M., Flaud J.-M. Joint ro-vibrational analysis of the HDS high resolution infrared data // J. Mol. Spectrosc. – 2006. - Vol.238. - P.23-40.
- Юхник Ю.Б., Бехтерева Е.С. Исследование колебательно-вращательного спектра поглощения молекулы AsD3 в районе 1350 - 1700 см-1 // Известия Томского политехнического университета. – 2006. – № 2 – С. 160–164.
- Юхник Ю.Б., Бехтерева Е.С., Синицын Е.А., Булавенкова А.С. Определение потенциальной функции молекулы AsH3 на основе экспериментальных данных // Известия Томского политехнического университета. – 2006. – № 4 – С. 61–65.
- Bekhtereva E. S., Ulenikov O. N., Albert S., Bauerecker S., Hollenstein H., Quack M. High resolution spectroscopy and dynamics of the methane molecule: CH2D2 isotopomer // Abstr. 61th International Symposium on Molecular Spectroscopy. – 2006. Columbus, Ohio, USA. - P.TB02.
- Синицын Е.А., Бехтерева Е.С., Булавенкова А.С., Улеников О. Н. О прецизионном определении структурных и динамических характеристик молекулы селеноводорода на основе экспериментальных данных // Известия вузов. Радиофизика. – 2007. – № 9 – С. 1-10.
- Бехтерева Е.С. Исследование колебательно – вращательного спектра молекулы CH2D2 в области 2000 – 3100 см-1 // Известия вузов. Физика. – 2007. Деп. в ВИНИТИ 23.11.2007 г. № 1091-В 2007.
- Bekhtereva E. S., Ulenikov O. N., Sinitsin E. A., Albert S., Bauerecker S., Hollenstein H., Quack M. On the semi-empirical determination of the methane potential energy surface // Abstr. 20th Colloquium on High Resolution Molecular Spectroscopy. – 2007. Dijon, France. - P.J30.
- Ulenikov O. N., Bekhtereva E. S., Leroy C., Gromova O. V., Flaud J.-M. On the SPGF approach in asymmetric top molecules: application to the hydrogen sulfide // Abstr. 20th Colloquium on High Resolution Molecular Spectroscopy. – 2007. Dijon, France. - P.M11.
- Ulenikov O. N., Bekhtereva E.S., Gromova O. V., Homiak T. V., Brger H. Ro-vibrational analysis of the ground and v3, v4, and v6 bands of AsH2D // J. Mol. Spectrosc. – 2008. - Vol.248. - P.286-293.
- Sanzharov N. A., Leroy C., Bekhtereva E.S., Ulenikov O. N. On the study of the vibrational energies of arsine molecule // J. Mol. Spectrosc. – 2008. - Vol.247. - P.1-24.
- Улеников О. Н., Бехтерева Е.С.., Леруа К., Громова О. В. Об определении потенциальной функции многоатомной молекулы: H2S // Известия вузов. Физика. - 2008. - № 1 - С. 17-22.
- Бехтерева Е.С. Об определении потенциальных функций H2CO, PH3 и CH4 на основе экспериментальных данных // Известия вузов. Физика. – 2008. – № 2 – С. 21-27.