

Синтез соединений ряда пиридазин-3(2 н )-она
На правах рукописи
ЧЕРКАЛИН Михаил Сергеевич
СИНТЕЗ СОЕДИНЕНИЙ РЯДА ПИРИДАЗИН-3(2Н)-ОНА
02.00.03 – Органическая химия
АВТОРЕФЕРАТ
диссертации на соискание ученой степени
кандидата химических наук
Ярославль – 2013
Работа выполнена на кафедре органической химии Федерального государственного бюджетного образовательного учреждения высшего профессионального образования «Ярославский государственный технический университет»
Научный руководитель: доктор химических наук
Колобов Алексей Владиславович
Официальные оппоненты: Шапошников Геннадий Павлович
доктор химических наук, профессор
ФГБОУ ВПО «Ивановский государственный химико-технологический университет»,
Смирнов Алексей Владимирович
доктор химических наук, доцент
Институт проблем хемогеномики ФГБОУ ВПО «Ярославский педагогический университет
им. К. Д. Ушинского»
Ведущая организация: ФГБОУ ВПО «Ярославский государственный университет им. П. Г. Демидова»
Защита состоится « 26 » сентября 2013 г. в 14.00 часов на заседании диссертационного совета Д 212.308.01 при ФГБОУ ВПО «Ярославский государственный технический университет» по адресу: 150023, г. Ярославль, Московский пр-т, д. 88, аудитория Г-219.
С диссертацией и авторефератом можно ознакомиться в научной библиотеке при ФГБОУ ВПО «Ярославский государственный технический университет» по адресу: 150023, г. Ярославль, Московский пр-т, д. 88.
Автореферат разослан «__» ___________ 2013 г.

Ученый секретарь диссертационного совета,
доктор химических наук А.А. Ильин
Актуальность темы. В настоящий момент значительная часть исследований в органической химии связана с поиском новых биологически активных структур и разработкой эффективных методов их синтеза. Азотсодержащие гетероциклические соединения в целом и пиридазины, как их представители, широко применяют в качестве лекарственных препаратов, регуляторов роста и средств защиты растений. Объем публикаций, посвященных исследованиям биологической активности этих соединений, в последние годы остается на высоком уровне. В настоящее время ведется разработка новых лекарственных препаратов позволяющих лечить сердечнососудистые расстройства, нарушения обмена веществ, заболевания нервной системы. Исследуются пиридазины которые позволяют бороться с диабетом и болезнью Альцгеймера. Поэтому получение новых соединений ряда пиридазина, изучение методов их синтеза и механизмов реакции, ведущих к их получению, является актуальной задачей. Настоящее исследование выполнено в соответствии с тематическим планом ЯГТУ, проводимым по заданию Федерального агентства по образованию РФ по теме: «Разработка методов синтеза ароматических, карбо- и гетероциклических полифункциональных органических соединений для получения композиционных материалов с использованием нанотехнологий» на 2008-2012 гг. (№ 0120.0 852836), а так же программой стратегического развития ГОУ ВПО ЯГТУ по теме «Материалы с новыми свойствами» 2012-2016 гг. (№0120 1275353).
Цель работы. Разработка методов получения новых производных 5- и 6-замещенных пиридазин-3(2Н)-онов, представляющих интерес в качестве билдинг-блоков для синтеза биологически активных веществ. Для достижения этой цели решались следующие задачи:
- Разработать методы синтеза новых пиридазин-3(2Н)-онов содержащих сульфонилхлоридную группу в 6-арильном заместителе, а так же сульфониламидов на их основе;
- Исследовать влияние структуры 6-арил-4,5-дигидропиридазинонов на реакцию образования пиридазин-3(2Н)-онов;
- Разработать методы получения новых эфиров и тиоэфиров 6-арилпиридазин-3(2Н)-онов;
- Разработать методы синтеза новых пиридазин-3(2Н)-онов на основе циклоалифатических кетонов и стероидных соединений.
Научная новизна. В ходе исследования взаимодействия замещенных 6-арил-4,5-дигидропиридазин-3(2Н)-онов с молекулярным бромом впервые показана зависимость относительной скорости дегидрирования от характера заместителя в арильном фрагменте, а так же заместителей в 4 и 5 положениях 4,5-дегидропиридазин-3(2Н)-онового фрагмента, предложен механизм реакции. Предложен новый однореакторный метод синтеза сульфонилхлоридов 6-арилпиридазинонов из 4,5-дегидропиридазин-3(2Н)-онов. Разработаны эффективные методы функционализации пиридазин-3(2Н)-онов, примененные для синтеза ранее неописанных соединений.
Практическая ценность. Разработаны методы синтеза новых соединений ряда пиридазина, в том числе стероидов, конденсированных в 16,17 положениях с пиридазин-3(2Н)-оном, являющихся, в соответствии с результатами расчёта потенциальной биологической активности, перспективными билдинг блоками. Получены и идентифицированы более 30 новых пиридазин-3(2Н)-онов. Разработан метод однореакторного синтеза сульфонилхлоридов 6-арилпиридазин-3(2Н)-онов, позволяющий увеличить выход целевых соединений.
Положения диссертации, выносимые на защиту: Влияние заместителей в арильном фрагменте 6-арил-4,5-дигидропиридазин-3(2Н)-онов на процесс дегидрирования под действием брома; механизм дегидрирования, включающий образование 5-бромпроизводного. Однореакторный способ получения сульфонилхлоридов 6-арилпиридазин-3(2Н)-онов. Методы синтеза сульфонилхлоридов, сульфониламидов, тиолов и стероидов, содержащих 6-арилпиридазин-3(2Н)-оновый фрагмент.
Апробация работы. Основные результаты исследования докладывались на всероссийской молодежной конференции-школе «Идеи и наследие А. Е. Фаворского в органической и металлоорганической химии XXI века» (Санкт-Петербург 2010); 63 научно-технической конференции студентов, магистрантов и аспирантов с международным участием, посвященной 1000-летию Ярославля (Ярославль 2010); международной конференции «Advance Science in Organic Chemistry» (Украина, Мисхор 2010); XIV молодежной конференции по органической химии. (Екатеринбург 2011); международной конференции «23rd International Congress of Heterocyclic Chemistry.» (Glasgow, UK, 2011), VI Всероссийской конференции молодых учёных, аспирантов и студентов с международным участием «МЕНДЕЛЕЕВ 2012» (Санкт-Петербург, 2012); 66 Всероссийской научно-технической конференции студентов, магистрантов и аспирантов высших учебных заведений (Ярославль 2013).
Публикации. По теме диссертации опубликовано 13 работ, в том числе 5 статей в российских журналах, включенных в список ВАК
Личный вклад автора заключается в постановке задач, разработке плана эксперимента, личном выполнении экспериментов, анализе и обобщении результатов, разработке представлений о механизме реакции, изложенном в работе, формулировании выводов.
Структура работы. Диссертация состоит из введения, литературного обзора, химической и экспериментальной частей, выводов, списка использованной литературы. Работа изложена на 116 страницах, включает 14 таблиц, 87 рисунков. Список литературы включает 122 источник.
Во введении определены актуальность работы, её цель, научная новизна и практическая значимость исследований, приведены основные положения, выносимые на защиту. Первая глава диссертации содержит обзор литературы по строению, способам синтеза, реакционной способности, и биологической активности соединений, содержащих пиридазиновый цикл, рассмотрены подходы к квантово-химическому моделированию данных систем. Во второй главе приводится обсуждение собственных результатов, идентификация ключевых соединений и выводы. В третьей главе содержатся характеристики исходных соединений, методики экспериментов и анализов.
Основное содержание работы.
Наиболее часто встречающимися прекурсорами 6-арилпиридазин-3(2Н)-онов являются 6-арил-4,5-дигидропиридазин-3(2Н)-оны. Эти соединения предполагают возможность их функционализации проведением реакций электрофильного ароматического замещения.
Для выбора наиболее подходящего пути получения замещённых в арильном фрагменте 6-арилпиридазин-3(2Н)-онов нами было рассмотрено поведение 6-арил-4,5-дигидропиридазин-3(2Н)-онов как в электрофильных реакциях, так и в реакциях дегидрирования.
1. Исследование дегидрирования 6-арил-4,5-дигидропиридазин-3(2Н)-онов под действием молекулярного брома.
6-Арил-4,5-дигидропиридазин-3(2Н)-оны способны дегидрироваться под действием различных реагентов, давая соответствующие 6-арилпиридазин-3(2Н)-оны. Благодаря высокой растворимости исходных реагентов, удобству выделения и высокому выходу ароматизация под действием молекулярного брома в уксусной кислоте является удобным и широко применяемым методом получения 6-арилпиридазин-3(2Н)-онов. Несмотря на это, механизм этого превращения, а так же влияние строения исходных 6-арил-4,5-дигидропиридазин-3(2Н)-онов на скорость реакции до настоящего времени не были исследованы.
Для установления зависимости реакционной способности 6-арил-4,5-дигидропиридазинонов от заместителя в арильном фрагменте, нами была использована реакция конкурентного дегидрирования.
6-Фенил-4,5-дигидропиридазин-3(2Н)-он, активность которого была принята за единицу, и исследуемое вещество в эквимолярных количествах вводили в реакцию с молекулярным бромом, взятым в недостатке. Методом жидкостной хроматографии оценивались молярные соотношения продуктов дегидрирования A/B (рисунок 1). Полученные таким образом значения соответствуют отношению констант скорости реакций для вещества сравнения и исследуемого вещества.

Рисунок 1 – Схема конкурентного бромирования 4,5-дигидропиридазинонов.
Полученные соотношения представлены в таблице 1.
Таблица 1 – Соотношение продуктов конкурентного бромирования (80°С, 2-5 мин, уксусная кислота, nисх a = nисх b = 0,0005 моль).
| Обозначение | R1 | Соотношение А/B, моль/моль |
| 1c | -OСН3 | 0,72 |
| 1b | -СН3 | 0,96 |
| 1a | -Н | 1,00 |
| 1d | -Cl | 1,08 |
| 1e | -COOH | 1,21 |
Анализ полученных результатов приводит к выводу, что усиление электронодонорных свойств заместителей в арильном фрагменте увеличивает активность соответствующих 4,5-дигидропиридазинонов. В литературе высказывается предположение, что превращение протекает через стадию образования 4-галогенпроизводного (Путь А). В этом случае процесс, очевидно, протекает по следующей схеме (рисунок 2).
Предложенный механизм не позволяет объяснить обнаруженную нами зависимость активности 4,5-дегидропиридазин-3(2Н)-онов от характера заместителя в арильном фрагменте, поскольку в этом случае ароматическое кольцо не может участвовать в стабилизации ни енола (2a-e), ни следующих за ним интермедиатов, в связи с отсутствием сопряжения.
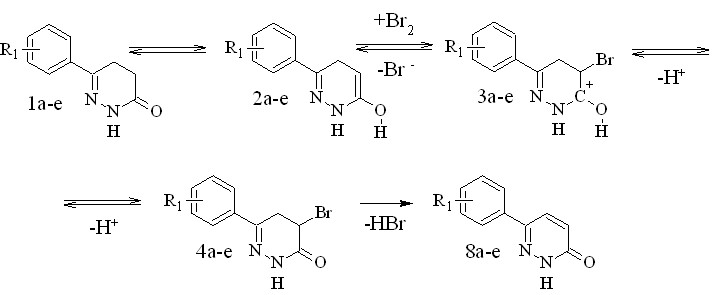
Рисунок 2 – Предполагаемый механизм дегидрирования 4,5-дигидропиридазинонов, включающий образование енола.
Можно предположить схему ароматизации, включающую стадию образования енамина с последующей электрофильной атакой двойной связи бромом (Путь B) в которой такое сопряжение присутствует (рисунок 3).

Рисунок 3 – Предполагаемый механизм дегидрирования 4,5-дигидропиридазинонов, включающий образование енамина.
Для определения наиболее предпочтительного направления реакции нами был произведен квантово-химический расчет как полуэмпирическими (AMPAC 9, PM6), так и неэмпирическими методами (Gaussian09, метод DFT). Расчет методом PM6 проводили как в газовой фазе, так и с использованием сольватационной модели COSMO для учета влияния растворителя на процесс. Оценивали теплоты образования исходных и конечных веществ, а так же предполагаемых интермедиатов.
Из анализа полученных данных можно сделать вывод о большей вероятности протекания реакции по пути B, поскольку енаминные формы соединений, а так же интермедиаты полученные из них в ходе реакции имеют меньшую энергию и являются более термодинамически выгодными по сравнению с енольными.
Для подтверждения полученных результатов был проведен расчет неэмпирическим методом DFT в приближении B3LYP с набором базисных функций 6-31G(d,p). Для учета растворителя была использована модель IPCM.
Как и в случае расчета методом PM6 полученные значения полных энергий интермедиатов пути B ниже чем в случае пути А, что позволяет предположить большую вероятность протекания реакции по пути B.
В таблице 2 представлены относительные изменения полной энергии в ходе элементарных стадий для обоих путей с учетом растворителя. В качестве нулевого значения приняты изменения полной энергии элементарных стадий превращения вещества 1a.
Таблица 2 – Относительные изменения полной энергии в ходе элементарных стадий. (DFT B3LYP 6-31G(d,p), IPCM, растворитель – уксусная кислота)
| Тип замести-теля | H-Ha, кДж/моль | |||||||
| Стадия превращения | ||||||||
| Путь А | Путь B | |||||||
| 1 2 | 2 3 | 3 4 | 4 8 | 1 5 | 5 6 | 6 7 | 7 8 | |
| a | 0,0000 | 0,0000 | 0,0000 | 0,0000 | 0,0000 | 0,0000 | 0,0000 | 0,0000 |
| b | 0,5403 | -4,3257 | 3,4696 | 0,4340 | 0,5427 | -6,3280 | 5,5712 | 0,3323 |
| c | 1,6451 | -10,1044 | 7,7599 | 1,5040 | 1,2943 | -14,7324 | 13,4105 | 0,8322 |
| d | -1,3057 | 5,9626 | -3,9496 | 0,0082 | -0,1115 | 9,1383 | -8,2129 | -0,0985 |
| e | -2,2772 | 10,9935 | -7,5745 | -0,4610 | -0,5339 | 15,1002 | -13,3926 | -0,4929 |
Для определения вероятной лимитирующей стадии данные тепловых эффектов элементарных стадий были соотнесены с натуральными логарифмами полученных значений относительных активностей. Как при использовании данных полуэмпирического расчета, так и неэмпирического было обнаружено наличие корреляционной связи для стадий электрофильного присоединения брома к енаминной форме исходного соединения, отрыва протона от образовавшегося при этом продукта и дегидрогаллогенирования. Поскольку процесс переноса протона не имеет барьера активации, а моногалогенпроизводных продуктов обнаружено не было, лимитирующей стадией, вероятнее всего, является электрофильная атака молекулой брома двойной связи. Для проверки этого предположения методом PM6 были найдены энергии переходных состояний и определены барьеры активаций (таблица 3). Структура переходных состояний была подтверждена решением колебательной задачи.
Таблица 3 – Энергии переходных состояний и барьеры активаций (RHF, метод PM6, COSMO, растворитель – уксусная кислота)
| a | b | c | D | e | ||
| H, кДж/моль | 2 | -171,69 | -217,29 | -350,60 | -211,71 | -551,15 |
| 2ПС | 1,35 | -42,46 | -178,16 | -40,22 | -382,21 | |
| Ea, кДж/моль | 173,04 | 174,83 | 172,44 | 171,49 | 168,94 | |
| H, кДж/моль | 5 | -214,09 | -259,37 | -395,34 | -254,12 | -593,53 |
| 5ПС | -57,91 | -104,06 | -243,00 | -96,52 | -434,31 | |
| Ea, кДж/моль | 156,19 | 155,31 | 152,35 | 157,60 | 159,22 | |
| Примечание: Буквами ПС обозначена структура переходного состояния образованная из соответствующего интермедиата | ||||||
Была определена зависимость натурального логарифма значений относительной активности от величины барьера активации (рисунок 4). Величина барьера для вещества сравнения была взята за ноль.
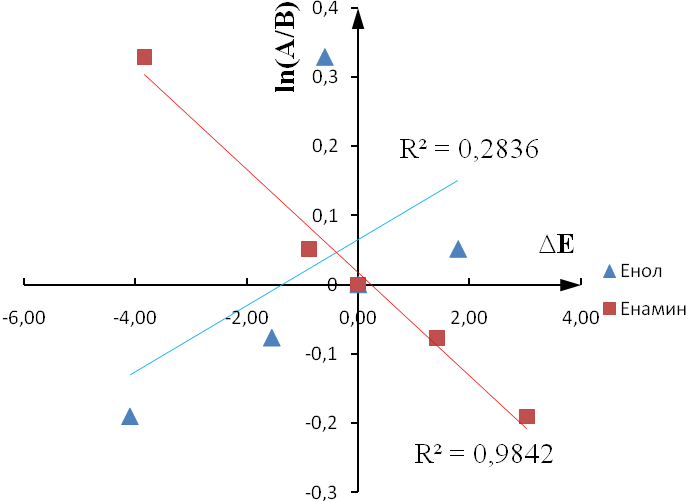
Рисунок 4 - Зависимость натурального логарифма значений относительной активности от величины барьера активации реакции присоединения брома к кратной связи
Полученные данные позволяют предположить, что процесс дегидрирования протекает по пути B, а стадия присоединения галогена может являться лимитирующей. В этом случае стабилизация образовавшегося карбокатиона за счет сопряжения -электронной системы ароматического кольца со свободной p-орбиталью положительно заряженного атома углерода является стабилизирующим фактором.
2. Разработка однореакторного метода синтеза сульфонилхлоридов 6-арилпиридазин-3(2Н)-онов.
Ранее было показано, что хлорсульфоновая кислота может выступать не только в роли сульфохлорирующего, но и как дегидрирующий агент. Полученные данные позволили предположить возможность однореакторного получения сульфонилхлоридов 6-арипиридазин-3(2Н)-онов из соответствующих 6-арил-4,5-дигидропиридазин-3(2Н)-онов в среде хлорсульфоновой кислоты без выделения промежуточных продуктов.
Ранее было обнаружено, что при реакции с хлорсульфоновой кислотой сохранение 4,5-дигидропиридазинон-3(2Н)-ового кольца возможно лишь в случае когда в арильном фрагменте присутствует сильный электронодонорный заместитель. Проведение реакции при повышенных температурах приводило к образованию смеси сульфонилхлоридов 6-арил-4,5-дигидропиридазин-3(2Н)-онов и продуктов их дегидрирования.
Учитывая закономерности, обнаруженные нами при дегидрировании под действием брома, можно предположить, что возможной причиной снижения реакционной способности является наличие электроноакцепторной сульфохлоридной группы. Нами были подобраны условия, при которых хлорсульфоновая кислота выступает в роли дегидрирующего агента на первой стадии, а затем как сульфохлорирующий агент на второй (рисунок 5).
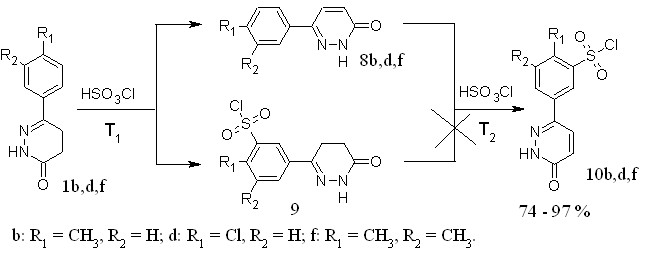
Рисунок 5 – Схема однореакторного синтеза сульфонилхлоридов 6-арилпиридазин-3(2Н)-онов.
Продукты реакции анализировали, переводя их в сульфамидные производные. В таблице 4 представлены условия проведения реакции и выходы полученных сульфонилхлоридов.
Таблица 4 – Условия проведения реакции взаимодействия 6-арил-4,5-дигидропиридазин-3(2Н)-онов с хлорсульфоновой кислотой и выход сульфонилхлоридов 6-арипиридазин-3(2Н)-онов (мольное соотношение субстрат : хлорсульфоновая кислота 1:11)
| Номер | Субстрат | T1,°С (, ч) | T2,°С (, ч) | Выход, % |
| 10b | 4-CH3 | 50°С (4ч) | 90°С (3ч) | 50 |
| 30°С (1ч) | 90°С (3ч) | 91 | ||
| 30°С (4ч) | 90°С (3ч) | 97 | ||
| 10d | 4-Cl | 40°С (3ч) | 140°С (4ч) | 90 |
| 10f | 3,4-CH3 | 30°С (3ч) | 90°С (3ч) | 74 |
Таким образом, предложен удобный способ получения сульфонилхлоридов 6-арилпиридазинонов без выделения промежуточных продуктов.
3. Синтез сульфонилхлоридов и сульфониламидов 6-арилпиридазин-3(2Н)-онов
Синтез новых пиридазин-3(2Н)-онов содержащих в своей структуре сульфонилхлоридную группу позволяет существенно расширить круг перспективных биологически активных соединений.
Как было установлено ранее, использование арилпиридазинонов, имеющих электронодонорный заместитель в четвертом положении бензольного кольца позволяет селективно проводить сульфохлорирование в третье положение.
Соединение 8g получали ацилированием бромбензола с последующей обработкой полученной 4-оксокарбоновой кислоты гидразин-гидратом и ароматизацией 6-(4-бромфенил)-4,5-дигидропиризазин-3(2Н)-она под действием брома. Для синтеза соединений 8h-k была использована схема включающая реакцию альдольной конденсации глиоксалевой кислоты с соответствующим ацетофеноном. Полученный продукт обрабатывали гидразин-гидратом. Соединение 8l было получено из 8i по реакции ацилирования ацетилхлоридом в пиридине.
Для синтеза сульфонилхлоридов соединения 8g-o прибавляли к избытку хлорсульфоновой кислоты (ХСК) (рисунок 6).
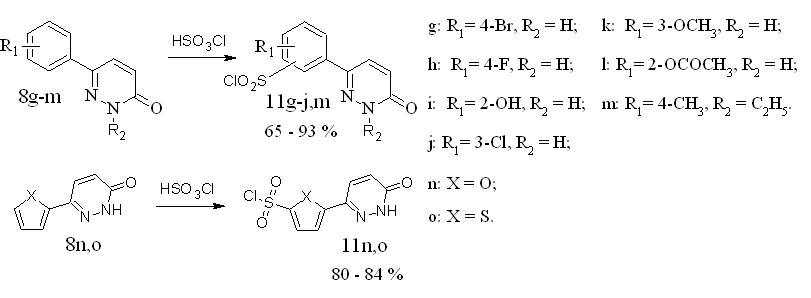 Рисунок 6 – Схема синтеза новых сульфонилхлоридов 6-арил и 6-гетерилпиридазин-3(2Н)-онов.
Рисунок 6 – Схема синтеза новых сульфонилхлоридов 6-арил и 6-гетерилпиридазин-3(2Н)-онов.
С целью увеличения выхода целевых продуктов реакцию проводили при различных соотношениях исходных реагентов, температурах и времени. В таблице 5 представлены условия реакций и выход целевых сульфонилхлоридов.
Таблица 5 – Сульфохлорирование пиридазин-3(2Н)-онов 8g-j,m-o
| Номер субстрата | Мольное соотношение ХСК:субстрат | T, °С | Время, мин | Выход, % |
| 8g | 11:1 | 140 | 150 | 93 |
| 8h | 6:1 | 140 | 210 | 70 |
| 8i | 6:1 | 25 | 150 | 65 |
| 8j | 8:1 | 100 | 300 | 70 |
| 8m | 6:1 | 80 | 180 | 92 |
| 8n | 6:1 | 50 | 300 | 84 |
| 8o | 6:1 | 50 | 300 | 80 |
Мольные соотношения хлорсульфоновой кислоты и субстратов соответствуют наибольшему выходу. Дальнейшее увеличение этого значения во всех случаях не приводило к увеличению выхода целевых сульфонилхлоридов.
Как видно из таблицы, в случае 6-(4-фторфенил)пиридазин-3(2Н)-она (8h) лучшие результаты были получены при прибавлении субстрата к 6-кратному избытку хлорсульфоновой кислоты при 20-25 °С, с последующим выдерживанием реакционной массы при 140 °С в течении 3,5 ч.
При сульфохлорировании 6-(4-бромфенил)пиридазин-3(2Н)-она (8g) наибольший выход был получен при проведении реакции с 11-кратным избытком хлорсульфоновой кислоты при 140 °С в течении 2,5 ч.
В отличие от арилпиридазинонов замещенных в четвертом положении арильного фрагмента при использовании 2- и 3-замещенных соединений региоселективность реакций определяется совместным влиянием заместителя и пиридазинового кольца. В некоторых случаях наблюдается образование смеси изомеров. Так, например, при сульфохлорировании 8k не удалось добиться получения индивидуального продукта. В остальных случаях были подобраны условия, приводящие к индивидуальному изомеру.
Лучшие результаты сульфохлорирования 8i были получены при прибавлении субстрата при 0-5 °С к 6-кратному избытку хлорсульфоновой кислоты с последующим выдерживании реакционной массы при 25 °С в течении 2,5 ч. Увеличение температуры привело к снижению выхода продукта 11i. Это, вероятно, связано с протеканием побочной реакции между сульфонилхлоридной группой одной молекулы и гидроксильной группой другой молекулы. Гидролиз данного продукта ведет к образованию водорастворимой сульфокислоты, которая теряется в процессе выделения. Повышение температуры может приводить к увеличению доли таких реакций и снижению выхода целевого сульфонилхлорида.
Было показано, что при сульфохлорировании 8l основным продуктом реакции является 4-гидрокси-3-(6-оксо-1,6-дигидропиридазин-3-ил)бензсульфонилхлорид (11i). По-видимому, ацильная защита подвергается гидролизу в процессе выделения. Несмотря на это атака, как и в случае 8i, протекает по пятому положению арильного фрагмента.
Проведение синтеза при 100 °С и увеличение времени реакции до 5 часов с 6-(3-хлорфенил)-пиридазин-3(2Н)-оном позволило получить чистый сульфонилхлорид 8j. Стоит отметить что продуктом реакции оказался 3-хлор-5-(6-оксо-1,6-дигидропиридазин-3-ил)бензсульфонилхлорид. Вероятно, причина такого необычного направления атаки заключается в стерической затрудненности орто- и пара- положения по отношению к хлору. В этом случае единственным доступным для атаки оказался углерод в мета- положении. Высокая температура и длительное время реакции привели к единственному продукту.
Сульфонилхлориды на основе 6-(фуран-2-ил)пиридазин-3(2Н)-она 8n и 6-(тиофен-2-ил)пиридазин-3(2Н)-она 8o были получены с наибольшим выходом при температуре 50 °С и времени реакции около 3,5 ч.
Для выделения полученных соединений реакционную массу небольшими порциями добавляли в ацетон, затем полученный раствор выливали в воду. Такой подход позволяет увеличить выход целевого продукта за счет снижения теплового эффекта, наблюдаемого при выделении реакционной массы на лед.
Из новых сульфонилхлоридов были синтезированы ранее неописанные сульфониламиды. Указанные соединения были исследованы в качестве потенциальных лекарственных препаратов и дали положительные результаты. Было показано, что эти вещества проявляют относительно высокую противовирусную активность при низкой токсичности.
4. Синтез новых серосодержащих пиридазинонов
Отработанные условия синтеза сульфонилхлоридов 6-арилпиридазин-3(2Н)-онов позволяют перейти к получению еще одного класса серосодержащих соединений – тиолов.
Последние можно получить на основе других серосодержащих производных (рисунок 7)
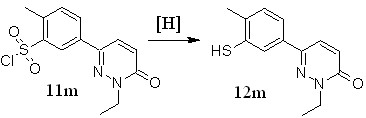
Рисунок 7 – Восстановление сульфонилхлоридов, содержащих пиридазиновый фрагмент.
В качестве восстановителей были выбраны хлорид олова (II) в тетрагидрофуране насыщенном хлороводородом и уксусной кислоте и трифенилфосфин в толуоле и тетрагидрафуране. Реакции проводили при 60°С в течение одного часа, затем отбирали пробу из реакционной массы и анализировали методом хромато-масс-спектрометрии. Восстановление хлоридом олова (II) не привело к образованию целевого продукта. При проведении синтеза в течение трех часов при температуре кипения растворителя был получен пиридазин-3(2Н)-он не имеющий заместителя в третьем положении бензольного кольца.
При использовании трифенилфосфина в тетрагидрафуране согласно данным масс-спектрометрии был получен целевой тиол 12m. Схожая картина наблюдалась при проведении реакции в толуоле.
Тиоэфирная группа может быть введена и до стадии образования пиридазинонового фрагмента.
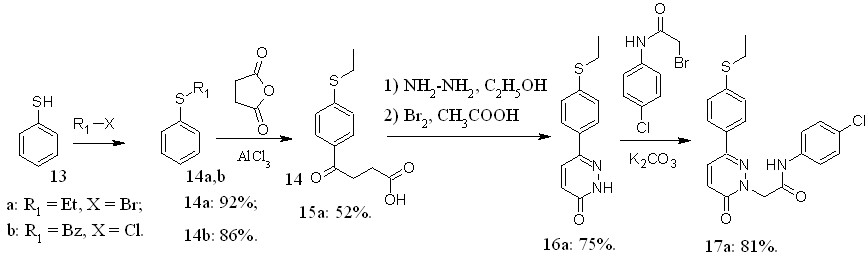
Рисунок 8 – Схема синтеза новых пиридазин-3(2Н)-онов, содержащих тиоэфирную группу.
Исходя из тиофенола (13) по схеме представленной на рисунке 8 нами был синтезирован ранее не описанный 6-[4-(этилсульфанил)фенил]пиридазин-3(2H)-он. На его основе была отработаны условия получения комбинаторной библиотеки N-замещённых производных, заключающиеся в проведении реакции в ДМФА с использованием карбоната калия при 80°С.
5. Синтез новых пиридазин-3(2Н)-онов на основе реакции альдольной конденсации.
Как отмечалось ранее, удобным способом получения 1,4-дикарбонильных соединений – прекурсоров пиридазин-3(2Н)-онов, является использование реакции альдольной конденсации.
На основе 2-гидроксиацетофенона, 2-ацетилфурана, 2-ацетилтиофена по реакции с глиоксалевой кислотой были получены соответствующие пиридазин-3(2Н)-оны (рисунок 9). Из них были синтезированы ранее не описанный N-алкил производные 18i,k,n,o.
 Рисунок 9 – Схема синтеза новых N-алкил производных пиридазин-3(2Н)-онов.
Рисунок 9 – Схема синтеза новых N-алкил производных пиридазин-3(2Н)-онов.
Реакции с циклоалифатическими кетонами - циклопентаноном (21a) и циклогексаноном (21b) протекает при более высокой температуре (рисунок 10).
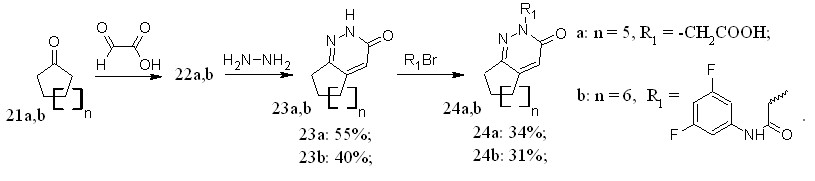
Рисунок 10 – Схема синтеза новых пиридазин-3(2Н)-онов на основе циклоалифатических кетонов.
На основе полученных циклоалифатических пиридазинов были синтезированы ранее не описанные N-алкил производные 24a,b.
Нами впервые была реализована схема получения пиридазинона 28 исходя из 2,3-бутандиона (рисунок 11).

Рисунок 11 – Схема синтеза (3,4-диметил-6-оксопиридазин-1(6H)-ил)уксусной кислоты из 2,3-бутандиона.
Отработанные условия синтеза пиридазин-3(2Н)-онов включающие реакцию альдольной конденсации позволили перейти к использованию в качестве исходных веществ стероидных соединений. Интерес к аналогам стероидных гормонов, конденсированных в 16,17 положениях с азотистыми гетероциклами, вызван свойственной им биологической активностью при отсутствии гормонального действия.
В качестве исходных соединений были использованы доступные 3-метоксиэкстр-1,3,5(10)-триен-17-он (29а) и 3--гидроксиандрост-5-ен-17-он (29b), которые вводили во взаимодействие с глиоксалевой кислотой, а затем обрабатывали гидразин-гидратом. Реакцию проводили как в условиях кислотного, так и основного катализа (рисунок 12).

Рисунок 12 – Синтез новых пиридазин-3(2Н)-онов, содержащих фрагмент стероида.
Образование целевых продуктов 3-метоксиэстр-1,3,5(10)-триен[17,16-c]пиридазин-31(2Н)-61-она 33a и 3--гидроксиандрост-5-ен[17,16-c]пиридазин-31(2Н)-61-она 33b возможно через образование промежуточных продуктов, соответсвенно, 30a и 30b с последующим спонтанным элиминированием молекулы воды (Путь А), либо через стадию кротоновой конденсации, приводящей к продуктам 32a и 32b (Путь В). В последнем случае становится возможным протекание побочной реакции присоединения гидразин-гидрата по активированной двойной связи. Поскольку данные продукты не были зафиксированы, мы предполагаем, что реакция протекает через стадию спонтанного элиминирования воды из соединений 31a и 31b (Путь А). Среди продуктов реакции были также обнаружены продукты взаимодействия гидразина с двумя молекулами карбонилсодержащих стероидов - азины, соответственно, 34a и 34b, образующиеся по схеме на рисуноке 13.

Рисунок 13 – Получение азинов из исходных стероидов.
В таблице 6 представлены использованные растворители, условия синтеза и состав выделенных продуктов.
Таблица 6 – Влияние условий проведения реакции на состав продуктов.
| Исходный стероид | Растворитель | Катали-затор | Время реакции (стадия 1) | Выделенные продукты | Выход, % |
| 1 | 2 | 3 | 4 | 5 | 6 |
| 29a | CH3COOH | - | 8 ч* | 33a, 34a | 10 |
| C2H5OH | K2CO3 | 16 ч | 29a | - | |
| KOH | 20 ч* | 33a, 34a | 30 | ||
| CH3CN | п-ТСК | 20 ч | 29a | - | |
| 39b | CH3COOH | - | 8 ч | 29b, 35 | - |
| C2H5OH | K2CO3 | 16 ч | 29b | - | |
| KOH | 20 ч* | 33b, 34b | 20 | ||
| CH3CN | п-ТСК | 20 ч | 29b | - | |
| * - проводили реакцию с гидразин-гидратом без выделения промежуточных продуктов (время реакции 2 ч., температура 100 °С) | |||||
При проведении реакции в условиях катализа карбонатом калия в этаноле после выдерживания реакционной смеси в течение 16 часов не наблюдалось образование промежуточных соединений 30a и 30b. После удаления растворителя были количественно выделены исходные стероиды 29a и 29b, соответственно.
Такой результат может быть связан со снижением электрофильности глиоксалевой кислоты вследствие образования соли и недостаточной основностью карбоната калия. Замена растворителя на ацетонитрил и использование п-ТСК не привели к желаемому результату.
При нагревании 29a в течении 8 часов при 110 °С в ледяной уксусной кислоте с 3хкратным мольным избытком глиоксалевой кислоты удалось выделить целевой продукт 33a с общим выходом 10%. Такой результат связан с низкой конверсии исходного стероида, что подтверждается обнаружением в продуктах реакции соединения 34a.
Проведение реакции в тех же условиях с субстратом 29b не приводит к образованию целевого продукта. В этом случае из реакционной массы был выделен продукт ацилирования гидроксильной группы - 3--ацетоксиандрост-5-ен-17-он.
При использовании 29a в присутствии гидроксида калия был выделен продукт 33a с выходом 30 %. Остальную часть составили упомянутый выше 34a и неидентифицированные смолообразые продукты.
Соединение 29b в тех же условиях реагирует с гораздо меньшей конверсией. Был выделен продукт 34b с выходом 68 % в расчете на исходный стероид. Целевое соединение 33b было получено с выходом 20 %.
Таким образом, наилучшие результаты были получены при использовании гидроксида калия в качестве катализатора в среде этилового спирта.
Выводы:
1. Показано, что наиболее вероятным механизмом реакции дегидрирования 6арил-4,5-дигидропиридазин-3(2Н)-онов под действием брома является механизм, заключающийся в преимущественном участии енаминной формы субстрата и образовании промежуточных 6-арил-5-бром-4,5-дигидропиридазин-3(2Н)-онов, подвергающихся в условиях реакции спонтанному дегидрогалогенированию. Предложенный механизм подтверждён экспериментальными данными и результатами квантово-химических расчётов.
2. На примере синтеза 6-[4-(этилсульфанил)фенил]пиридазин-3(2H)-она показана возможность получения тиоэфиров ряда 6-арилпиридазин-3(2Н)-она исходя из тиофенола. Определены условия восстановления сульфонилхлоридной группы в 6-арилпиридазин-3(2Н)-онах в тиольную заключающиеся в проведении реакции в толуоле с использованием трифенилфосфина.
3. Установлено, что проведение реакции 6арил-4,5-дигидропиридазин-3(2Н)-онов с хлорсульфоновой кислотой с выдерживанием реакционной смеси в течении 1-4 часов при температуре 30-50 °С, а затем 90-140°С в течении 3-4 часов приводит к получению сульфохлоридов ряда 6-арипиридазин-3(2Н)-она.
4. Показано, что исходя из 3-метоксиэкстр-1,3,5(10)-триен-17-она и 3--гидроксиандрост-5-ен-17-она можно получить ранее неизвестные 3-метоксиэстр-1,3,5(10)-триен[17,16-c]пиридазин-31(2Н)-61-он и 3--гидроксиандрост-5-ен[17,16-c]пиридазин-31(2Н)-61-он, представляющие интерес в качестве аналогов соединений обладающих ярко выраженной биологической активностью при отсутствии гормонального действия.
5. На примере 2,3-бутандиона впервые показано, что альдольная конденсация о-дикарбонильных соединений с эфирами малоновой кислоты и последующее взаимодействие продуктов реакции с гидразингидратом приводит к 5, 6 замещённым пиридазин-3(2Н)-она.
Основное содержание работы изложено в следующих публикациях:
1. Черкалин М. С. Сульфонилхлориды арилпиридазинонов и сульфониламиды на их основе / М. С. Черкалин, Т. А. Бобова, А. В. Колобов // Изв. Вузов, Химия и хим. технология, - 2012. - том. 55, №. 11, C. 13-16.
2. Черкалин М. С. Синтез производных пиридазинона из ароматических и циклоалифатических кетонов / М. С. Черкалин, А. А. Шетнев, Т. А. Бобова и др. // Изв. Вузов, Химия и хим. технология, - 2011. - том. 54, №. 12, C. 12-14.
3. Овчинников, К. Л. Взаимодействие 6-арил-4,5-дигидро-3(2Н)-пиридазинонов с молекулярным бромом. Особенности и механизм реакции / К. Л. Овчинников, М. С. Черкалин, А. М. Курманов и др. // Изв. Вузов, Химия и хим. технология, - 2011. - том. 54, №. 9, C. 36-38.
4. Бобова, Т. А. Алкилирование 4-R-замещенных-2Н-фталазин-1-онов / Т. А. Бобова, А. А. Шетнев, М. С. Черкалин и др. // Изв. Вузов, Химия и хим. технология, - 2011. – том 54, № 11. – C. 41-43
5. Бобова, Т. А. Сопряженное присоединение азотсодержащих гетероциклических соединений к производным малеиновой и итаконовой кислот / Т. А. Бобова, А. В. Колобов, М. С. Черкалин и др. // Изв. Вузов, Химия и хим. технология, - 2012. - том. 55, №. 8, C. 3-5.
6. Zavarzin, I. V. Synthesis of steroid [16,17]pyridazin-3-ones / I. V. Zavarzin, M. S. Cherkalin, A. V. Kolobov et al. // Материалы международной конференции «23rd International Congress of Heterocyclic Chemistry.» 31 July - 4 August 2011, Glasgow, UK, p. 140-142.
7. Черкалин, М. С. Синтез серосодержащих производных 6- арилпиридазин-3(2H)-онов / М. С. Черкалин // Материалы VI всероссийской конференции молодых учёных, аспирантов и студентов с международным участием «МЕНДЕЛЕЕВ 2012», Санкт-Петербург, 2012. C. 501-502.
8. Черкалин, М. С. Синтез новых производных пиридазин-3(2Н)-она / М. С. Черкалин, А. В. Колобов // Материалы XIV молодежной конференции по органической химии. Екатеринбург. 2011. C. 50.
9. Пат. 245504 РФ. Способ получения сульфохлоридов ряда 6-арилпиридазин-3(2H)-онов / А.М. Курманов, К.Л. Овчинников, М. С. Черкалин, А. В. Колобов, Е. Р. Кофанов, Я. В. Садовникова // Заявлено 01.04.11.
10. Черкалин, М. С. Синтезы на основе 3,6-дихлорпиридазина / М. С. Черкалин, С. С. Рожков, К. Л. Овчинников и др. // Материалы международной конференции «Advance Science in Organic Chemistry». Украина, Мисхор 2010,
C. 56.
11. Черкалин, М. С. Синтез и свойства 6-(2-гидроксифенил)-пиридазин3(2Н)-она / М. С. Черкалин, С. С. Рожков, К. Л. Овчинников // Материалы 63 научно-технической конференции студентов, магистрантов и аспирантов с международным участием посвященной 1000-летию Ярославля, Ярославль 2010, C. 342.
12. Черкалин, М. С. Особенности синтеза дигидропиридазинонов, содержащих норборненовый фрагмент / М. С. Черкалин, А. В. Колобов // Сборник тезисов «Всероссийская молодежная конференция-школа «Идеи и наследие А. Е. Фаворского в органической и металлоорганической химии XXI века. Санкт-Петербург 2010, C. 143.
13. Монастырева, Д. А. Синтез новых сульфидсодержащих пиридазин3(2Н)онов / Д. А. Монастырева, М. С. Черкалин, А. В.Колобов // Материалы 66 всероссийской научно-технической конференции студентов, магистрантов и аспирантов высших учебных заведений с международным участием, Ярославль 2013, C. 440.