

Генетические основы этиологической гетерогенности злокачественных новообразований
на правах рукописи
КАЗУБСКАЯ ТАТЬЯНА ПАВЛОВНА
Генетические основы этиологической
гетерогенности злокачественных новообразований
14.01.12 - онкология
03.02.07 - генетика
Автореферат
диссертации на соискание ученой степени
доктора медицинских наук
Москва 2010 г.
Работа выполнена в Учреждении Российской академии медицинских наук, онкологическом научном центре им. Н.Н.Блохина РАМН
(Директор академик РАН и РАМН, профессор М.И.Давыдов)
Научные консультанты: доктор медицинских наук, профессор
Раиса Федоровна Гарькавцева,
доктор медицинских наук, профессор,
Владимир Юрьевич Сельчук.
Официальные оппоненты: доктор медицинских наук, профессор
Петр Васильевич Новиков,
член-корреспондент РАМН,
доктор медицинских наук, профессор
Николай Евгеньевич Кушлинский,
доктор медицинских наук, профессор
Валентин Сергеевич Мазурин.
Ведущая организация: ГОУ ДПО Российская медицинская академия постдипломного образования Минзравсоцразвития России
Защита диссертации состоится « » 2010 года в часов на заседании диссертационного совета (Д.001.017.02.) по адресу:115478 Москва, Каширское шоссе, д.24.
С диссертацией можно ознакомиться в библиотеке РОНЦ им.Н.Н.Блохина РАМН.
Автореферат разослан « » 2010г.
Ученый секретарь диссертационного Совета,
доктор медицинских наук, профессор Ю.А.Барсуков
Общая характеристика работы ӯ Актуальность темы.
Интерес клиницистов к роли генетиӯчесӯких факторов, как первичных факӯтоӯров риска в этиологии рака поӯвыӯшался с последоӯваӯтельӯными открыӯтиями герӯмиӯнальӯных мутаций генов Rb, BRCA1/2, RET, ассоциированных с раӯзвиӯтиӯем ретиноӯблаӯсӯтомы (Friend SH. et al.,1986, Lee WH.et al.,1987), рака молочӯной жеӯлезы (Woӯoster R. at al.,1994, Ford D. at al.1994), синдромов множестӯвеӯнӯӯӯных энӯдоӯкӯринӯных неоплазий 2 типа (МЭН2) (Takahashi M.еt al.,1985). Инӯтеӯӯрес стал более интенсивным, когда выяснилось, что анализ клиниӯчесӯкой карӯӯтины болеӯзни, выявление связи между клиниӯчесӯкиӯми проявленияӯми заӯбоӯлевания и лежащими в их основе молекуляӯрӯными повреждеӯниями, отӯкӯрыӯваӯӯют возӯможӯности идентиӯфиӯкаӯции новых форм болезӯни и установления приӯчин их возникноӯвеӯния, что может привести к более нацеӯленному изменению раӯкового контӯроӯля и таргетӯной терапии.
С генеӯтичесӯкой точки зрения главной особенностью неоплазий является клинический полиморфизм и генетическая гетероӯгенӯноӯсть, а для так назыӯваӯемых мультифакториальных злокачественных опухолей - суӯщеӯствоӯваӯние их наӯследственных и ненаследственных форм без четких граӯниц. Проблема геӯнеӯӯӯтичесӯкой гетерогенности наӯследственных форм опухолей занимает важное место, поскольку кажущееся феноӯтипичеӯски однородным, генетически гетеӯрогенное заболевание, может включать в себя несколько клинически, генетиӯчески и биоӯхимически самостоятельных форм (Гинтер Е.К.,2001). Анализ клиниӯчесӯӯкой картины болезни (фенотипа) и выявление взаимосвязи между генотипом и фенотипом, преӯӯдоӯставляет возможность установить не только какие генетиӯчеӯсӯкӯие измеӯнеӯӯӯния лежат в основе формирования предрасполоӯженности к неоӯплазии, но и разработать стратегию формирования прицельно однородӯных групп риска, опӯтимизиӯроӯвать процедуру проведения ДНК-анаӯлиза. Изуӯчение особенӯностей проявления неоплазий и причин, которые их форӯмиӯруӯют, являӯется одним из важнейших направлений в медиӯциӯӯне вообще и клиниӯчесӯкой генеӯтике в частности, и лежит в основе успешной диагӯносӯтиӯки и медиӯко-генеӯтического консультирования больных и их семей.
Указанные осоӯбенӯности характерны и для рака щитовиӯдной железы (РЩЖ), мелаӯномы кожи (МК) и первично-множественӯных злокачестӯвенных опухолей (ПМЗО). Имеюӯщӯиеся на сегодӯняшний день работы, посвященные гетерогенӯности этих форм опухолей, не имели сисӯтемӯного подхода и ограниӯчиӯвались областью профессиоӯнальӯных интереӯсов (моӯлекулярно-генетичесӯкиӯми изучеӯниями отдельных нозоӯлогичесӯких форм неоӯпӯлазий). Однако недоӯсӯтаӯӯӯточная изученӯноӯсть отдельӯных генетиӯческих вариантов злокачественных опуӯхоӯлей может привести к тому, что в рамках одной нозоӯлоӯгиӯчесӯкой формы рака, моӯгут расӯсматӯриваться заӯбоӯлеӯвания с различӯной генеӯтиӯчеӯской приӯроӯдой и различными патогенетическими механизмами развития.
Проӯбӯлема своевременӯного выявления РЩЖ, МК, ПМЗО закӯлюӯчается не только в бесӯсимптомном развитии, но и в отӯсутствии наӯдежӯных марӯкеров, способствующих их раннему выявлению. Наряду с этим, перӯвично-мноӯӯжесӯтӯвенӯные опухоли предстӯаӯвӯляет собой особую диагӯносӯтичеӯсӯкую проӯбӯлему. Изуӯӯӯчеӯния, прицельно направӯленӯӯные на выявление спеӯциӯфиӯӯческих приӯзнаков, котоӯрые могли бы у пациенӯтов увеличивать риск проявӯлеӯӯния вторых первичӯных раков, являются актуӯаӯльӯными с клинической и биолоӯгической точек зреӯния. Значение проблемы возрастает еще и в связи с неуклонным ростом заболеӯваӯемости и смертӯности от этих новоӯобразований (Давыдов М.И., Аксель Е.М., 2009). В этой связи осоӯбое значение имеет иденӯтификаӯция наӯсӯледӯстӯвенӯных форм РЩЖ, МК, ПМЗО, разработка стратегии преодоӯления генеӯтиӯческой гетероӯгенноӯсти с исӯпольӯзованием биологических параӯметров на клиниӯчесӯком, геӯнетичеӯском и молеӯкуӯӯӯляӯрӯном уровнях, которая позӯвоӯлит иденӯтифицироӯвать семьи со схоӯдӯными клиническими признаками. При этом активӯӯное выявӯлеӯние даже относительно небольшого числа случаев злоӯкачесӯтвенӯӯных новоӯобӯраӯзований среди практиӯчесӯки здоровых родственников из сеӯмей больных этими забоӯлеӯваниями моӯжет рассматриваться как важная науӯчно-практиӯчесӯкая задача.
И хотя оценка геӯнеӯтичесӯких параметров насӯледӯстӯвенных форм опухолей затӯруднена, решеӯние этих вопросов имеет первоӯсӯтеӯӯӯпенное значение для разӯработки прогӯрамӯмы клинико-генетической диӯаӯгӯноӯстики, профиӯлактики (в том числе и преӯнаӯтаӯӯльной) и патогенетически ориӯентированной терапии. Все эти вопросы плаӯниӯровались и разработаны на моӯдеӯли рака щитовидной жеӯлеӯзы, меланомы кожи, перӯӯвичӯно множестӯвенных злоӯкаӯчеӯсӯтвенных опухоӯлях.
Цель и задачи исследования.
Цель работы: определить роль наследственных факторов в этиолоӯгии рака щитовидной железы, меланомы кожи и первично множестӯвенных злокачестӯвенных опухолей, выделить генетически детермиӯнироваӯнӯӯные формы этих заболеваний, изучить их генетическую гетерогенӯность, разӯработать тактику медико-генетического консультиӯроӯвания при разных их ваӯриӯантах.
В соответствии с поставленной целью были сформулированы следующие задачи:
1. Изучить размах вариабельности клинико-генетического проявления рака щитовидной железы (РЩЖ) меланомы кожи (МК) и первично множестӯвенӯных злокачественных опухолей (ПМЗО).
2. Определить фенотипические и генетические корреляции различных фоӯрм опухолей в семьях больных РЩЖ, МК и ПМЗО.
3. Изучить клинические, цитогенетические и молекулярно-генетические особенности РЩЖ, МК и ПМЗО.
3.1 Изучить частоту и спектр молекулярных изменений при медуллярном РЩЖ (МРЩЖ).
3.2 Определить частоту и спектр повреждений некоторых участков хромоӯсом в клетках крови больных МК и диспластических невусов (ДН).
3.3 Изучить молекулярно-генетические и эпигенетические изменения гена р16/CDKN2A в образцах опухолей МК.
3.4 Оценить значимость биологических микрочипов в диагностике геӯрӯмиӯнальных мутаций генов BRCA1,2 и CHEK2 у больных первично множеӯсӯтвенӯным раком с поражением яичников.
3.5 Изучить статус метилирования генов-супрессоров опухолевого роста: RASSF1A, RAR2, SEMA3B, опредеӯлить их значение в молекулярном патоӯгеӯнезе и диагностиӯке солитарного и первично мноӯжесӯтвенного рака молочной железы и яичников.
4. Установить структуру и соотношение генетически детерминированӯных форм РЩЖ, МК и ПМЗО.
5. Разработать основные приӯнципы медико-генетичесӯкого консультироваӯния семей РЩЖ, МК и ПМЗО.
Научная новизна и практическая значимость
Впервые на основе комплексного клинико-генетического и молекулярно- биологического изучения больных раком щитовидной желеӯзы, меланомой кожи и первично множественными злоӯкачественными опухолями выявлены основные клинико-генетические особенности этих заболеваний. Расширены представления о генетической геӯтеӯроӯгенности (алӯлельного или локусного проӯисхождения) РЩЖ, МК, ПМЗО, определена структура и соотношение их генетически детерминированӯных форм, разработаны критеӯрии для выӯделения генетически самосӯтоӯӯятельных вариантов.
Показано, что молекулярӯной основой МЭН2 являются мутации в гене RET, установлена четкая ассоциация муӯтаӯций в этом гене с синдромами множестӯвенных эндокринных неоплазий 2 типа, определен спектр его мутаций, обнаӯружены новые мутации гена RET у росӯсийских больных, что позволяет расӯширить спектр мутаций, который можно рекомендоӯвать для рутинного лабоӯраторного анализа. Установлено, что приӯчиной клиниӯческого поӯлиӯӯморӯфиӯзма медуллярного РЩЖ является разное положение точкоӯвой мутации в одном из цисӯтеӯиӯӯноӯвых кодонов гена RET, обнаружиӯвая аллеӯльӯӯную гетерогенӯность этого заӯболевания. Впервые в стране по результаӯтам ДНК-диагносӯтики меӯдуӯллярного РЩЖ проӯвеӯдено проӯфилакӯтиӯчесӯкое удаление щитовидӯной желеӯзы, внеӯдӯрена в праӯкӯтиӯку пренаӯтаӯльӯная диагӯносӯтика этого заболеваӯния. ӯӯӯӯӯӯӯ Разработана комплексная программа выявӯлеӯния лиц предрасположенӯных к развитию МРЩЖ, позвоӯляющая не только утоӯчӯнить индивидуальный генеӯтический диагноз, дать более точный прогӯноз теӯчеӯния заболевания, но и осуӯщеӯӯстӯвить своевременные лечебно-профилактиӯческие мероприӯятия, направӯленӯные на предотвращение развития этой опухоли. ӯ ӯӯӯ
Выявлено наличие характерных для МК и ДН генетических и эпигенетиӯчесӯких нарушений. У индивидов с МК, МК+ДН и у лиц носителей диспласӯтиӯческих невусов в трех участках короӯтӯкого плеча хромосомы 1-1р22,1р31, 1р32 наблюдается общӯноӯсть в инӯдивиӯдуӯальном распреӯдеӯлеӯнии раӯзрывов и отличается от нормы с высоӯкой достоӯверӯностью, что ассоцииӯруется с локальӯной хромоӯсомӯӯной нестаӯбиӯльностью и является ранним событием в цепи изӯмеӯӯӯнений, предшествующих МК. Аномальное метилирование проӯмоторного райӯона гена CDKN2A является неслучайным изменением у больных МК и выӯявляется у всех больных ПМЗО с поражением МК.
С использоваӯнием технологии олигонукӯлеоӯтидных биоӯчипов показано, что частота наиболее распространенных герминальных мутаӯций в гене BRCA1 у больӯных ПМЗО с поражением яичниӯков выявӯляӯется в 53% случаев.
Впервые проведено определение статуса метилирования СрG-островков геӯнов RASSF1A, RAR2 и SEMA3B в опухолевых и нормальных тканях молоӯчӯной железы, яичников больных первично множественными опухолями, больӯных соӯлитарным РМЖ и РЯ и лимфоцитах периферической крови здоровых доноров. Показана высокая частота аномального метилирования промоторӯных районов этих генов в тканях больных ПМЗО с поражением молочной жеӯлезы и яичниӯков и солитарных РМЖ и РЯ, с частотами от 30% до 90%. Впервые показано, что боӯльные с аномальным метилироӯванием генов RASSF1A, RAR2 могут иметь тенденцию к первично множественӯному пораӯжению РМЖ и РЯ. Выяӯвӯӯлена досӯтоверная положительная корреляция часӯтоӯты метиӯлирования генов RAR2 и SEMA3B со стадией и степенью анаплазии РМЖ и РЯ. Поӯкаӯӯзаӯно, что аномальӯное метилирование промоторной области гена RASSF1A можӯно обнаӯруӯжить на доклинической стадии развития этих опухолей, что позвоӯляет рекоӯмендовать их в качестве молекулярных маркеӯров диагностики и прогноза этих заболеваний и свидетельӯсӯтвует о больших возможностях меӯтилирования ДНК для разработок неинваӯзивӯной диагносӯтики рака.
Внедрение в практику.
Результаты исследования внедрены и используются в клинической практике различных подразделений РОНЦ им.Н.Н.Блохина РАМН.
Апробация работы.
Материалы диссертации представлены и обсуждены на Российсӯких онкоӯлогиӯчесӯӯких конгрессах:VII,X,XI,XII (Москва 2003, 2006-2008 г.); научно-праӯӯкӯтиӯчесӯкой конференции "Пробӯлеӯмы онкоӯгенеӯтики: научӯные и приӯклаӯдӯные асӯпекӯты", Украина, Киев, 2002; Conference on Frontiers in Canӯcer Prevention Reӯsearch (USA, Boston, 2002), научно-практической конференӯции НИИ онӯкоӯлогии ТНЦ СО РАМН (Томск, 2003); Conference on Frontiers in Cancer Preӯvenӯtion Research (Toronto, 2003); Human Genome Meeting“HGM'99”( Scotӯland, Edinburgh, 2001); 28th Meeting of the Federation of Euroӯpean Biocӯhemica Sociӯeties (Turkey, Istanbul, 2002); Российской конференции по онкогинекоӯлоӯгии (Москва, 2009); European School of Oncoloӯgy “Меланоӯма” (Москва 1995), “Thyӯroid cancer” (Moscow, 2004); cъезде онкологов ресӯпуӯблики Казахстан (Алма-Ата, 2003); I Всероссийӯском съезде онкологов «Совӯременӯные техӯноӯлогии в онколоӯгии» (Ростов-на-Доӯну, 2005); IV съезде онкоӯлогов и радиоӯлоӯгов СНГ (Баку, 2006); конӯфереӯнӯции с межӯдуӯнародным учаӯсӯтием "Генетика в Росӯсии и мире" (Москӯва, 2006); 30th Conference of American Society of Preӯventive Oncology (USA, Bethesda, 2006); 4-й Российской конӯференции по фунӯдаӯментальной онкологии “Петӯровские Чтения”, (Санкт-Петербург, 2008); 19th Internatiӯonal Congӯress on Anti Cancer Treatment (ICACT) (Paris, France, 2008); III конӯгрессе с междуӯнародным участием «Опуӯхоли головы и шеи», (Сочи, 2009); II Росӯсиӯйском симпозиуме «Молеӯкулярӯно-геӯнеӯтическая диагӯностика злокачесӯтӯвенных опухолей человека» (Москва, 2009).
Апробация работы состоялась 11 декабря 2009 года на совместной научной конференции с участием лаборатории клинической онкогенетики, отделения биотерапии опухолей, отделения гинекологичесӯкого, отдеӯлеӯния опухолей молочных желез, проктологического отделения, отдела опухоӯлей головы и шеи НИИ клинической онкологии РОНЦ им. Н.Н. Блохина РАМН; а также кафедры генетики и кафедры онкологии факультета послеӯдиӯпломного образования Московского государственного медико-стоӯмаӯтолоӯгиӯческого университета Министерства здравоохранения и социального развиӯтия.
По теме диссертации опубликовано 83 печатных работы в российских и заӯрубежных журналах, из них – 36 в журналах, рекомендуемых ВАК МОН РФ, 3 работы оформлены как главы в справочниках и учебных пособий.
Структура и объем диссертации. Диссертационная работа изложена на 245 страницах машинописного текста и состоит из введения, обзора литераӯтуры, материалов и методов, трех глав собственных результатов исследоӯвания и их обсуждения, выводов, списка литературы, который содержит 22 отечественных и 290 зарубежных источников. Работа илӯлюстӯрирована 39 таблицами, 2 графиками, 52 рисунками.
Материалы и методы исследования.
В НИИКО РОНЦ им.Н.Н.Блоӯхина РАМН за 1988-2008 годы сформирован канӯӯцер-реӯгистр лаборатории клинической онкогенетики, cтавший осноӯвой исӯӯӯӯслеӯдуемой выборки, объем которой составил 1495 больӯных и 8700 их родӯстӯвенӯников 1-2-й степени родсӯтва. Из них 208 больных РЩЖ (140 папилӯлярӯным и фолӯлиӯкулярным РЩЖ и 68 больӯных медуӯлӯлярӯным РЩЖ), 460 боӯльӯных МК, 400 больных ПМЗО, жителей Москвы и облаӯсти. Каждая семья реӯгистрироӯваӯлась через одного члена семьи, т.е. путем одиӯноӯчӯӯного отбора. Особое вниӯмание удеӯлялось подтверждению диагноза онколоӯгиӯӯчесӯкоӯго заӯболевания у роӯдӯственӯниӯков I степени родства: родителей, сибсов (братьӯев и сестер), деӯтей, которые затем включались в дальнейший анализ. ӯ ӯ ӯ ӯ ӯ ӯ ӯ
В анализе испольӯзоӯвались клинические, эпидемиологические, клиӯӯниӯӯко-геӯӯнеаӯлоӯӯгичесӯкие, цитоӯгеӯнетиӯческие, молекуӯлярно-генетические, геӯнеӯӯтико-маӯтеӯматиӯчеӯсӯкие методы исӯследования. У всех пациӯентов было полуӯчено инфорӯмиӯрованное согласие об участии в исслеӯдованиӯях. Все случаи рака классифиӯцироӯваны по TNM согласно требованиям Международного противоракового соӯюза (UICC, версия 1989 г.)
В качестве контроля использовались популяционные частоты распростӯраӯненӯности разӯличных злокачественных новообразований, которые были выӯчисӯлены по данным учетӯных форм № 090/V посӯтуӯпивӯших из всех районных онкоӯлогических отделений г. Москвы в отдел медицинской статистики МГОД. Данӯные о половозрасӯтном составе насеӯления г. Москвы в изучаеӯмый периӯод быӯли получены в Московском городском статистическом упраӯвлеӯнии. Покаӯзатели заболеӯваемости были стандартиӯзованы по возрасту пряӯмым методом с испольӯзоӯванием мирового стандарта возрастного распредеӯлеӯния населения.
Идентификация наследственных синдӯромов проводилась в соответствии с усӯтаӯновленными междуӯнародӯными критериями для каждого из них.
Изучение распӯроӯстӯраӯненности ДН в г. Москве проводилось на основании резульӯтатов осмотра 509 человек, случайно отобранӯного континӯгенӯта взросӯлого насеӯления. Для поиска специфических опухолевых ассоциӯаций в семьях больных МК исӯӯпольӯзоваӯлись клинико-генеалогические данные, включавшие 98 челоӯвек имеӯӯюӯщих на коже мноӯжесӯтвенные меланоцитарные невусы и их 259 родстӯвенӯников 1-й стеӯпени родства и 22 человека с мноӯжественными невусами, отоӯбӯранных при осмотре 247 близких родственӯников из семей 91 больӯного с гисӯтоӯлоӯгиӯчески подтвержденным диаӯгӯнозом МК. Каждый пациӯент-носитель множественных неӯвусов получил лечеӯние и наблюӯдался от 5 до 18 лет в клиӯнических отделеӯниӯях РОНЦ в пеӯриӯод с 1988 по 2005 г. В качесӯтӯве контрольӯных групп слуӯжили 2 выӯборки: одна из них состояла из 1001 болӯьного с гисӯтоӯлоӯгически подтвержӯденӯным диагноӯзом - рак желудка и 3793 их родстӯвенӯника 1-й стеӯпени родӯства; втоӯрая включала 1048 клиниӯчески здоӯровых лиц, не имевӯших злокаӯчественных опуӯхолей и их 4654 родстӯвенника 1-й степени родства также собӯранӯных среди мосӯковского населения в период с 1985 по 2003 годы. Все анализируӯемые групӯпы были подобраны по полу и воӯзӯрасту и были жителями Москвы и Московӯсӯкой области.
Молекулярные исследования, ДНК-диагӯносӯтика наличия точкоӯвых мутаӯций в протоӯонӯкоӯӯгене RET проводилась на базе Медико-генетиӯчесӯкого научӯного центӯра РАМН совместно с лабоӯраторией молекуӯлярной генетики и клиӯӯникой Гамӯбургӯского униӯверсиӯтета. Материалом для исследоӯваний служиӯла ДНК пациӯентов медуӯлӯлярным РЩЖ находившиеся в РОНЦ им.Н.Н.Блохиӯна РАМН с 1998 по 2007г. Геномную ДНК выделяли из лимӯфоцитов перифеӯрической крови, обӯразӯцов опуӯхоӯлеӯвых и нормальӯных ткаӯней щиӯӯтоӯӯвидной железы (кусочки или гисӯтолоӯгические срезы) и испольӯзовали в качестве матриӯцы для ПЦР-ампӯлиӯфиӯкаӯции экзонов гена RET.
Цитогенетическое изучение наиболее частых перестроек встречающихся при МК, вклюӯӯчаюӯщих в себя сайты повыӯшеӯнӯной ломкости хроӯмоӯсом(СПЛХ), конституӯтиӯвные (кСПЛХ) или fragile sites проӯвоӯдили на G-окраӯшенных меӯтаӯӯфазных пластинах лимфоӯциӯтов перифериӯчеӯской крови, кульӯтиӯвируемых по стандартной метоӯдике, проводилось совместно с лабораторией цитогенетики в РОНЦ им.Н.Н.Блохина РАМН. Молекулярно–генетические исслеӯдоӯваӯния МК проӯводиӯлись в лаборатоӯрии эпигенетики Медико-генетичеӯского наӯучӯноӯго центӯра РАМН. Материалом исследования служила ДНК лимӯфоциӯтов пеӯриферичесӯкой крови и ткани опухолей больных МК. Для исследоӯваний исӯпоӯльӯзовали 10 микросателлитных маркеров на хромосоме 9р, распоӯложенӯных в райӯоне гена р16/CDKN2A. Анализ метилирования проӯмоӯторной облӯасӯти геӯна р16/CDKN2A был проведен методом метилчувствитеӯльӯной ПЦР.
Определение спектра и частоты наиболее частых точковых мутаций в геӯнах BRCA1/2 и CHEK2 у больных первично-множественӯными опухолями с пораӯжеӯнием яичников и больӯных органоӯспециӯфичесӯким раком яичников в поӯпуляции российӯских женщин проводились на базе Инстиӯтута молекуӯлярӯной биологии им. В.А.Энгельдгардӯта. Методом аллель-специӯфичной гибриӯдиӯзаӯции, сочетаӯюӯщего муӯльтиплексную ПЦР и гибриӯдизацию с олигонукӯлеоӯӯтидӯными биочипами, опӯреӯдеӯлялась часӯтота встречаемости мутаций в гене BRCA1- 185delAG, 300T>G, 4153delA, 4158A>G и 5382insС, в гене BRCA2- 695insT, 6174delТ и в гене CHEK варианта 1100delC.
Исследование уровня метиӯлироӯвания промоторных районов геӯнов RASSF1A, RAR2, SEMA3B проводилось на базе Государственного Научӯного Центра РФ ГосНИИ генетика и совместно с Каролинӯским инӯӯстиӯтуӯтом (Швеӯция). Исслеӯдоӯваӯӯние включало комбинацию методов: ПЦР-анаӯӯӯлиз образцов геӯноӯмной ДНК, посӯледовательно расщипленной с помощью набора метилӯчувӯствительных рестӯрикӯтаз (МЧРА), бисульфитной модификации ДНК с посӯледующей метилспеӯциӯфиӯчӯной ПЦР. В анализ включены 330 пар обӯраӯзӯцов ДНК, выделенной из опуӯхоӯлеӯвой и гистологиӯчеӯски нормальной ткани боӯльӯных раком молочной железы, яичников и первично-множественӯныӯми опухоӯлями с поражением молочной железы и яичӯниӯӯков. В качестве конӯтӯӯроля исӯпоӯӯльӯӯзовали лимфоӯциты перифериӯческой крови здороӯвых индиӯвидов (15 челоӯвек). ДНК выдеӯляли из ткани опуӯхоли и из лимфоӯцитов крови по стандартӯной метоӯдиӯке.
Переӯсмотр гистологических препаратов проведен в отделе патоморфолоӯгии РОНЦ им. НН.Блохина РАМН.
Исследование проведено с использованием генетико-математических меӯтоӯдов изложенных в методических разработках (Трубников В.И., Москва, 1998), в пакете программ “Biometrika”. Для создания базы данных испоӯльӯзоӯвался прогӯрамӯмный продукт фирмы VNIINSoft КАРАТ. Основные стаӯтисӯтиӯческие методы – критерии Фишера, ранговая корреляция Спирмана с испольӯзоӯваӯнием t-теста Стьюдента и критериев.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Проведенное исследование посвящено одной из сложных проблем в клиӯниӯческой онкологии: изучению генетической обусловленности и генетиӯчеӯсӯкой гетерогенности и клинического полиморфизма рака щитовидной желеӯзы, мелаӯӯноӯмы кожи и первично-мноӯжественных злокачественных опухолей.
Клинико-генетический анализ рака щитовидной железы. ӯ ӯ ӯ Раннее выявление РЩЖ, клинико-генетическая идентификация его насӯлеӯдӯӯстӯвенных варианӯтов, разработка методов выявления аналогичного забоӯлеӯваӯния среди родстӯвенӯниӯков больных РЩЖ и тактика лечения бессимпӯтоӯмӯных ноӯсиӯтелей патоӯлогиӯческих мутаций гена/ов, ответственных за разӯвиӯтие РЩЖ, являӯеӯтӯся важӯной задачей онкологии и генетики. Хотя папилляӯрӯный и фолӯлиӯкулярӯный (неӯмеӯдуллярный) РЩЖ (НМРЩЖ) является наиӯболее часӯтой форӯмой рака этой локализации, о его наследӯстӯвенӯном эквиваӯлеӯнӯте изӯвеӯсӯтӯно очень мало.
для решения вопроса о наследственной предрасположенӯности к НМРЩЖ быӯли изуӯчены 140 больных, у которых папиллярный рак набӯлюӯдалӯся у 74%, папилӯлярно-фолӯликуӯлярный в 23% и в 3% слуӯчаев - фолликуӯлярный. При анаӯӯлизе установлены различия в проявӯлеӯнии этого заболеӯваӯӯния в заӯвиӯӯсиӯӯӯмоӯсти от пола (соӯотноӯшение мужӯчин и жеӯнӯщин -1:5), что указывает на суӯщеӯсӯтӯвоӯвание разӯличных величин риска развития НМРЩЖ для мужчин и жеӯнӯщин. Возӯраст диӯаӯгностики заболеваӯния в среднем составляет 47,7 ұ1,61 года, у 78% боӯльӯӯных перӯвые признаки заболевания проявляются в среднем на 6 -7 лет раньӯше. У боӯльӯных с НМРЩЖ обнаӯруӯжено сочетанное поражеӯние разӯных органов, частота первиӯчӯно-мноӯжесӯтвенӯных неоплаӯзий составила 5,7%, что выше часӯтоты в попуӯляции (0,003%)(р0,01). Анализ показал наӯлиӯчие сеӯмейного наӯкопӯления разных неоплазий, частота которых составила 4% и добӯроӯкачественных опухолеӯвых и неопухолевых заболеваний щиӯӯӯтоӯвиӯдной желеӯзы - составила 3,2%.. ӯӯ При изучении семейных случаев НМРЩЖ впервые установлен наследсӯтӯвеӯнӯный синдром семейного папиллярного РЩЖ (ПРЩЖ), признаками котоӯрого явӯляӯются ӯпреимущесӯтӯвенно мультифоӯкаӯльӯное и двустороннее поражеӯние паӯпилӯлярным раком только ткани щиӯтоӯвидӯной жеӯлезы у трех и более члеӯнов сеӯмьи, рис.1. Возӯраст диагностики ПРЩЖ оказаӯлся мноӯго моӯложе общей выӯборӯки больӯных НМРЩЖ, составив 34,1ұ 6,8 года против (47,7 ұ 1,6 года) (р 0,05), в цеӯлом, показыӯвая боӯлее тяжеӯлый фенотип и аутосомно-домиӯнантный тип (роӯдиӯтели-дети) насӯлеӯдоӯваӯния заӯбоӯӯлеӯваӯния.
Родословная семей больных С и В, диагноз: синдром семейного ПРЩЖ
I
II
III
Рис.1. Отец больной (II-2) умер в 36 лет от ПРЩЖ, (II-1) умер в 38 лет от ПРЩЖ, (III-2) –папилӯлярный РЩЖ, его бабушка (I-4) умерла от ПРЩЖ на фоне множественӯных аденоӯмаӯтозных узлов щитовидной железы, а (I-5) –умерла в 73 года от рака поджелудочной железы.
В результате исследования выявлены наследственно обусловленные синӯдӯроӯмы, компонентом которых является папиллярный и фолликулярный РЩЖ. Эти синдромы характеризуются двумя клинико-генетическими ваӯриӯантами. Пеӯрвый, ассоциирован с множественными типами опухолей, возӯниӯкаӯющих в ткаӯнях раӯзӯных органов и наследуеӯмых по аутоӯсомӯно-домиӯнантӯноӯму типу. Это сиӯнӯдромы -Гардӯнеӯра, Каудена и МЭН1, которые в анаӯлиӯзируӯеӯмой выӯборӯке больных идентиӯфиӯциӯроӯваны в 3,6% случаӯев. Второй - синдром семейного ПРЩЖ(СПРЩЖ), при котоӯром преобладаюӯщим и постоӯянным призӯнаком является паӯпиллярӯный РЩЖ, обнаруӯжен в 1,4% слуӯчаев.
Проведенное исӯследование показало существование насӯледӯсӯтӯвенно обусӯлоӯвӯӯлеӯнных форм НМРЩЖ и в отдельно взятых семьях коӯсегреӯгаӯцию НМРЩЖ с неоӯплазиями почки, моӯӯлоӯчӯной железы, кожи (мелаӯноӯмы), нервӯной системы и толсӯтой киӯшки. В целом, среди больных НМРЩЖ наследӯственно детермиӯнированӯные форӯмы этого заболевания выявлены в 5% слуӯчаев.
Полученные данные позволяют иденӯтифицировать семьи с наследственно обуӯсловленными формами НМРЩЖ и формиӯроӯвать группы "высокого рисӯка" среӯди родственников из семей больных НМРЩЖ, а такӯже осуществлять индивиӯдуӯальный прогноз развития заболевания для членов семьи больӯноӯго в зависимосӯти от конкретной семеӯйӯной ситуации. На основании полученной информации, разӯраӯботаны критерии идентиӯфиӯкации лиц с высоким рисӯком развития НМРЩЖ, которые включают: 1) пораӯжение НМРЩЖ до 40 лет и наличие пораӯженных члеӯнов семьи аналоӯгичным заболеванием; 2) наличие в анамнезе или в сеӯмье друӯӯгих неоӯпӯлазий: в почӯках, толстой кишке, молочной железе, нервной сисӯтеӯмы, кожи и наличие гаӯмарӯтом; 3) наличие в анамнезе или у членов семьи доброӯкаӯчеӯсӯтвенӯных опуӯхолей в щитовиӯдной железе; 4) наличию в сеӯмье ПМЗО с поӯраӯӯжением щитоӯвидной железы; 5) внимание к щитовидной жеӯлеӯзе при идеӯнтиӯфиӯкаӯции у паӯциентов наследсӯтӯвенных мульӯтиӯопухолевых синдӯромов.
Важной проӯблемой является выявление С-клеточной гиперплазии или меӯдуӯлӯлярного РЩЖ (МРЩЖ) на ранней стаӯдии заболевания. В этой связи, доӯстижеӯния в молекуӯлярной биологии, позвоӯлили не только понять причиӯны развития медулӯлярӯного рака, но и реально поӯӯдойти к решению вопроса ранӯней диагносӯтики и профилактики этой форӯмы РЩЖ.
Проведенный анализ 68 семей больных МРЩЖ выявиӯл генетически деӯтеӯрӯминированӯный МРЩЖ в 24,9% случаев, которые имели разные формы проӯявления, такие как МЭН2А, МЭН2Б, сиӯндром СМРЩЖ и медуллярный РЩЖ как компонент нейӯӯроӯфиброматоза 1 типа. Исӯхоӯдя из важного наблюӯдеӯния, что ген RET экспӯрессируется только в опреӯдеӯлеӯнных тканях нейроэнӯдокӯринӯной дифӯӯференӯциӯровки, включая параӯфоллиӯкуӯляӯрные С-клетки щитоӯвидӯной железы, экстра и интраадренаӯлоӯвые хромаӯфиӯнӯные клеӯтки надпочечӯниӯков, периӯфериӯческие нервы, описываемые случаи совӯместӯного поражеӯния этиӯӯми редкиӯми формами забоӯлеваний как нейрофибӯроӯматоз и МРЩЖ предӯполаӯгаӯют возможӯность существования обӯщего биоӯлоӯгичесӯкого мехаӯниӯзма, ответӯстӯвенӯного за их развитие. Частота спорадиӯческого МРЩЖ сосӯтаӯвила 75,1%, среди коӯторых, моӯгут быть случаи обусӯлоӯвӯӯленные мутацией "de noӯvo" в полоӯвых клетках одного из родиӯтелей.
При изучении семей с синдромами МЭН 2 типа усӯтаноӯвлен широкий спеӯӯктр клиничесӯкого проявления МРЩЖ, высокая вариаӯбеӯльность риска раӯӯӯӯзӯӯӯӯвиӯтия неоӯплазии среди родственников. Для синдрома МЭН2А характерно биӯлаӯтеӯральӯное разӯвитие феохромоцитом у больных и их родӯстӯӯвенников, коӯтоӯрые являлись частой причиной внеӯзапӯной смерти. Наличие феохромоӯциӯтоӯмы усӯтаӯӯновӯлено в 50% семей, у одного из больных, помимо МРЩЖ и феӯоӯхроӯмоӯциомы, был обнаружен треӯтий первичный рак молочӯной железы. Выявӯлена асӯсоциӯация МРЩЖ с аноӯмаӯлией почек, обнаруӯженная у носиӯтельницы муӯтации гена RET.
Анаӯлиз семей с синдромом МЭН2Б показал, что 90% боӯльных является реӯзульӯтатом мутации de novo. МРЩЖ при этом синдроме возникает гоӯраӯздо раӯньше (как правило, до 5 летнего возраста) и протекает более агресӯсиӯвно. Для синдроӯма семейного МРЩЖ характерно поражение родственӯниӯков сайт-спеӯциӯфичесӯким медуллярным раком при отсутствии клиниӯчесӯких или биохиӯмичеӯсӯких призӯнаков пораӯжеӯния паращитоӯӯвиӯӯдных желез или надӯпоӯӯчечӯников.
Возраст манифестации МРЩЖ у пациентов и их родстӯвенӯников с МЭН2 ваӯрьирует в зависимости от метода диагностики. При МЭН2А возраст диаӯгносӯтиӯки в среднем 28 лет (колебался от 7 до 45 лет), а у неӯӯӯкоторых родстӯвенников к моӯменту изучения этих семей возраст превыӯшал 50, 62 и 75лет. При СМРЩЖ он в среднем составил 30 лет, в то время как при МЭН2Б развитие заболеӯвания наблюӯдаӯлось в среднем 8,6 года, покаӯзыӯвая самый тяӯжелый фенотип.
Впервые в России проведен скрининг герминальных мутаций в гене RET в семьях больных с МЭН2. В результате установлена четкая ассоциация муӯтаӯций в этом гене с МЭН2 синдромами. В семьях с МЭН2А идентифициӯроӯваны 2 спеӯцифиӯческие муӯтаӯции в кодоне 634 гена RET, в семьях с МЭН2Б выӯяӯвлена мутаӯция в кодоне 918 гена RET. При синӯдӯроме сеӯмейного МРЩЖ обнаӯружена муӯтаӯция в кодоне 620 гена RET. Хаӯраӯкӯтерӯные для этих синдӯромов межӯиндиӯвидуӯальная изӯмеӯнӯӯчивость, как по возӯрасту наӯчала забоӯлеваӯния, так и по тяӯжеӯсти его прояӯвӯлеӯния (даже в преӯдеӯлах одӯного подӯтипа) могут быть обусӯловлеӯны не только варьӯируӯющей экспӯрессивӯноӯстью и пенеӯтӯранӯтӯностью гена RET, но и положением точковой муӯтаӯции в одном из цисӯтеӯиновых кодонов этого гена. Молекулярная ДНК-диаӯгностика гена RET, стаӯла реӯальным подтверӯжӯдеӯнием выявлеӯнных наследсӯтвенӯных форм МРЩЖ.
Налиӯчие аллельной гетерогенности находит свое отражение в выявӯленӯной взаӯӯимосвязи между феӯноӯтипом- RET генотипом и тяжестью проявления заӯболеӯваӯния. У больных с MЭН2A муӯтаӯция в кодоне 634 ассоциӯируется с феоӯхроӯмоӯцитомой и гиперпаратиреозом и до сих пор, не быӯла обнаруӯжеӯна ни в одной из семей с сиӯнӯдромом семеӯйӯного МРЩЖ, что позволяӯет рассматӯриӯвать ее в качеӯсӯӯӯтӯве марӯкеӯра при проӯгӯнозиӯровании риӯска развития этих забоӯлеваний у бесӯсимӯптомных носиӯтелей этой мутаӯции. Геӯрӯмиӯнаӯльные мутации в кодоне 918 хаӯраӯктеӯрӯны искӯлючиӯтельно для МЭН2Б и свяӯзаӯны с пороками развития скелета и слиӯзиӯстых. Выявленная ассоциация позволяет дать более точный прогӯноз течеӯния заболевания у больных МЭН2.
Единственным эффективным метоӯдом лечения МРЩЖ является тиреӯоидӯэкӯтоӯмия, поэтому так важна ранӯняя и доӯклиӯническая диӯаӯгностика этого заболеӯвания. Однако вопрос о профилактической тиреоидэктомии остается предӯмеӯтом посӯтоӯӯӯянной дискуссии, поскольку все еще отсутстӯвуют данные длиӯтеӯльӯного наблюӯдеӯния за пациентами, перенесшими в раннем детском возрасте тиреоидӯэкӯтомию, осӯнованӯную на прямом тестировании гена RET, и которые могли бы покаӯзать, что МРЩЖ радикально излеӯчен. Прямое теӯстирование гена RET среди родстӯвенӯӯников больных МЭН2А обӯӯнаружило 8 бессимпӯтомӯных носитеӯлей герминаӯльӯных мутаӯций этого гена в возрасте от 3,5 до 42 лет из 4-х обсӯледоваӯнных нами семей (табл.1). Первый российский опыт профиӯлаӯкӯтиӯчесӯкого хиӯӯӯӯруӯӯрӯгичесӯкого лечеӯния выӯполӯнен у 6 из них, согласившихся на опеӯӯративное лечеӯние. Как оказалось, тольӯко у двух из них, самых юных пациӯенӯтов (3,5и 9 лет), опеӯраӯция была проӯӯӯфилактиӯчесӯкой, у остальных обӯнаӯружеӯны очаги С-клеӯтоӯчӯной гиперӯплаӯзии и медуллярный рак.
Таблица 1.
Клиническая характеристика родственников семей с МЭН 2А и результаты тестирования гена RET (выявленный генотип) каждой семьи.
| Пациенты: № семьи, (возраст/пол) | Наличие феоӯхроӯмоцитомы/ медулӯлярӯного рака у паӯциенӯтов ( Ф/М) | Мутация гена RET (генотип) замена аминокислоты/кодон | Бессимптомные носители мутации гена RET (+ ), тиреоидэктомия |
| Семья №1: 32 года/м 50 лет/м 3,5 года/ж 9 лет/м 38 лет/ж 9 лет/ж 10 недель, беременности | ф/м ф - - - - - | TGCCGC,коӯдон 634, экзон 11 Не тестировался TGCCGC, коӯдон 634, экзон 11 ( - ) ( - ) ( - ) ( - ) | (+ ) тиреоидэктомия |
| Семья №2: 35 лет/ж 25 лет/ж 29 лет/ж 20 лет/м 9 лет/ж 2 года/м | ф/м ф ф м - - | TGCCGC,коӯдон 634, экзон 11 Не тестировалась Не тестировалась TGCCGC, коӯдон 634, экзон 11 TGCCGC, коӯдон 634, экзон 11 ( - ) | (+ ) тиреоидэктомия (+ ) тиреоидэктомия |
| Семья №3: 14 лет/ж 21 год/ж 42 года/ж 31 год/ж 69 лет/ж | М - М Не обследована Визуальное увеӯличение щитоӯвиӯдной железы | TGCCGC, коӯдон 634, экзон 11 ( - ) TGCCGC, коӯдон 634, экзон 11 TGCCGC, коӯдон 634, экзон 11 TGCCGC, коӯдон 634, экзон 11 От обследоваӯния и лечения отказалась | (+ ) тиреоидэктомия ( + ) ( + ) |
| Семья №4: 14 лет/ж 12 лет/м 38 лет/ж 74 года/ж | М М М щиӯтовид. железа удаӯлеӯна в 45 лет | TGCGGC,кодон634, экзон11 TGCGGC, кодон634, экзон11 TGCGGC, кодон634, экзон11 Не тестировалась, от обследоваӯния и лечения отказалась | (+ ) тиреоидэктомия (+ ) тиреоидэктомия |
Эти данӯные покаӯзаӯли, что носиӯтели мутаций могут быть подвергнуты тиреӯоидӯэкӯтомии до поӯяӯвӯления МРЩЖ. Послеопеӯраӯционное наӯблюӯдение от 1,5 до 8 лет (в средӯнем 4,8 года) позволило установить, что тиӯреӯӯоӯидэктомӯия проӯвеӯдеӯнная на доӯклиӯӯниӯчеӯском уровне, позвоӯляӯет изӯбеӯжать меӯӯтаӯстаӯзироӯваӯния и имеет боӯлее благоприӯятӯный исход заӯбоӯлеваӯния (табл.2), раннее выявӯление заӯбоӯлеваӯние на осноӯве ДНК-тесӯтиӯӯрования может изменить течение МРЩЖ.
Изучение исходного и стимулиӯрованӯного уровня каӯльцитонина - как диаӯгӯӯносӯтического маркера показало, что этот тест не всегда является абсоӯлютӯным криӯтерием. У пациентки 3,5 лет из семьи с МЭН2А набӯлюдаеӯмый уроӯвень кальӯӯӯциӯтоӯниӯна был выӯше нормы почӯти в 2,5 раза (76,8 пг\мл), одӯнаӯко при гисӯтолоӯгиӯчесӯком исӯслеӯдоӯваӯнии призӯнаков МРЩЖ обнаӯруӯжено не быӯло (табл.2). Неоспориӯмая ценноӯсть этого метода в возӯмоӯжӯности оцеӯнить полноӯту хиӯрурӯгиӯчесӯкого лечения.
Таблица 2.
Клиническая характеристика членов семей больных МЭН 2А, до и после
тиреоидӯэкӯтомии
| Данные обследования пациентов до тиреоидэктомии | ||||||
| Возраст | 3,5 года | 9лет | 14 лет | 20 лет | 38 лет | 42 года |
| Кальцито- нин (пг/мл) | 76,8 | 42,3 | 97,0 | 20,3 | 40,0 | 83,4 |
| Катехола-мины | норма | норма | Норма | норма | норма | норма |
| Мутация гена RET | + | + | + | + | + | + |
| Данные обследования пациентов после тиреоидэктомии | ||||||
| Гистолоӯгиӯ ческое ис-ӯ следование щитоӯвид. железы | Без приз- ӯнаков С-ӯ ӯ клеточ. гиӯперпӯӯлаӯзии | Без призӯна ков С-кле- точной ги- перплазии | С-клеточная гиперплазия, узлы медул- лярного рака (0,7-1,0 см) | Множ. узлы медулл. рака (0,7-1,0 см), очаги С-кл. гиперпл. в обеих долях | Множ. узлы медулл. рака (до1,0 см) с амилоидо-зом в обеих долях | Множеств. узлы медул- ляр.рака (до 1,0 см) на фоне С-клет. гиперплазии |
| Кальцито- нин (пг/мл) | норма | норма | норма | норма | 80,3 | норма |
| Катехол-амины | норма | норма | норма | норма | норма | норма |
| Длительно-сть наб-ния после опер. | 4 года | 8 лет | 3 года | 7 лет | 5 лет | 2 года |
Исӯӯӯхоӯдя из приӯобӯретенного опыта, пациӯенӯӯтам с мутаӯцией в кодоне 634, мы рекоӯмендуем провоӯдить тиреӯоидӯэкӯтоӯмию независимо от уровӯня кальӯциӯтоӯӯниӯна. Выяӯвӯӯленная взаимоӯсвязь генотиӯпа и фенотипа у носиӯтелей этих муӯтаций моӯжет способӯствовать плаӯнироӯваӯнию тиреоидэктомии, приӯнятию реӯшеӯния отӯноӯӯсительӯно объема и времени провеӯӯӯния профилакӯтичесӯкой тиреӯоидӯэкӯтоӯмии, разӯработӯке тераӯпевӯтиӯчеӯских подхоӯдов по отношеӯнию к пациӯентам-носиӯтеӯлям мутаций. Комбиӯнированный с молеӯкулярными техноӯлоӯгиӯями подӯход к клиӯниӯчеӯской, биоӯхиӯмичесӯкой диагӯностике и ведению пациӯенӯтов из сеӯмей с МЭН2А может стать основным в уменьӯшение летальӯности от МРЩЖ.
Первый опыт дороӯдоӯвой диагностики синдӯрома МЭН2 в нашей стране проӯӯӯӯвеӯден в семьях с МЭН2А и синдромом семейного МРЩЖ. Пренатальная диаӯгӯӯносӯтика проӯведеӯна в семье №1 (табл.1), где у паӯциента 32 лет с синдӯроӯмом МЭН2А (носителя мутаӯции в кодоне 634) ожидалось появӯлеӯние потоӯмства. На сроӯке беӯреӯменӯности 10 неӯдель проведена из клеӯток ворсин хоӯриӯона, у его будуӯщего реӯбенка, была выделена геномная ДНК и выполӯнена прямая ДНК-диагӯноӯсӯтика гена RET. Для исклюӯчеӯния контаминации исслеӯдуӯемого материӯала матеӯринӯӯсӯӯкиӯми тканями провеӯдено контӯрольӯное исследоваӯние. Мутаӯции гена RET не обнаӯруӯжено, выӯӯявлен мужӯской пол плода, роӯдиӯлся здороӯвый мальчик. В возрасте 1 меӯсяца, ему проӯвеӯдено повторное тестироӯвание ДНК, анализ окаӯзался отрицаӯтеӯльӯӯным. В сеӯмье с синдромом СМРЩЖ, у паӯциӯентки с мутаӯциӯей TGC(Cys)CGC(Arg) в коӯдоне 620 (экзон10), в 35 лет на фоне лечения МРЩЖ встал вопӯрос о дорӯоӯдовой диагӯностике. На сроӯке беӯреӯмеӯнӯности 13-14 недель провеӯдено прямое тестироваӯние гена RET, у плоӯда выявӯлена анаӯлоӯгиӯчӯная мутацияӯ гена RET. Пациентка приӯняла решение преӯрӯвать беременӯность.
Таким образом, приӯмеӯнеӯние RET-анализа дает возможӯность диаӯгӯностироӯвать (или искӯлюӯӯӯчить) наследственную форму МРЩЖ, подготовить роӯдитеӯлей к рожӯдению больӯноӯго ребенка или предотвраӯтить рождение такоӯвого.
Проведенный поиск соӯматичеӯсӯӯких мутаций в гене RET у больӯных со споӯраӯдическим МРЩЖ позвоӯлил идеӯнӯтиӯфиӯцировать мутаӯции в 28,6%. Выявӯлено 6 тиӯпов соматических мутаций, три из которых - гетероӯзиӯготные мутаӯции в кодоӯнах 639, 641 и 922 оказались неизвестными ранее. Это дает новые сведеӯния по молекулярной генетике синдромов МЭН2 и позволяет расшиӯрить спеӯктр мутаӯций, который можно рекомендоӯвать для рутинного лабоӯраторного анаӯлиза. При изучении мутаӯций гена RET в образцах крови 13 больных со споӯраӯдичесӯким МРЩЖ, обнаӯружена гермиӯнаӯльӯная муӯтаӯӯция “de novo” тем саӯмым показана неоӯбходиӯмость ДНК-тестиӯрования споӯрадичесӯких форм МРЩЖ. Созданы предпоӯсылки для формирования банка геӯнеӯтической инӯфоӯрӯмаӯции клинически верифиӯцированных больных с МЭН2 в России.
На основе полученных в ходе исследования данных, высокий риск развиӯтия гиперӯпараӯтироза и феохроӯмоцитомы в семьях с синдроӯмом МЭН2А преӯӯӯдӯӯӯполаӯгает необходимость включения в скриниӯруюӯщие прогӯраммы диаӯӯгӯносӯтиӯку этих заболеваний. Выявление муӯтаӯции в кодоне 918 геӯна RET свиӯдеӯтеӯльӯствует о саӯмом агрессивном типе синдромов – МЭН2Б и воӯӯӯзӯможӯӯноӯсӯти раӯнӯнего меӯӯтаӯсӯтазиӯрования заболеӯвания и соотӯветӯственно плаӯниӯроӯвания возӯӯраста тиреоӯидӯэкӯтоӯмии, выӯбор котоӯрого провоӯдится индивидуӯаӯльно, но не позднее первых 5 лет жизни. Синӯдӯром сеӯмеӯйӯӯного МРЩЖ проӯтеӯкает менее тяжело и возникает в более позӯдӯнем возрасте. ДНК-диагностику в семьях с МЭН2 слеӯдует проводить осноӯвыӯваӯясь на вариӯантах этого синдӯроӯма и в соответӯсӯтвии с хараӯктерисӯтиӯками мутаций гена RET.
Структура и соотношение генетически детерминированных форм МРЩЖ вклюӯчает: 1) семейный синдром МЭН2А (7,4%); 2) МЭН2Б (11,7%); 3) синӯдӯром СМРЩЖ (4,4%); 4) в составе синдӯрома Реклингхаузена (1,4%).
Проведенный генетический анализ МРЩЖ позволил разработать компӯлеӯксную программу выявления лиц, предраспоӯложенных к развитию этого заӯбоӯлевания, включающую три этапа мероприятий:1) выявление и реӯгисӯтӯраӯӯция сеӯӯмей с MЭН2; 2) ДНК-диагностика бесӯсимптомӯных членов сеӯмей с наследӯсӯтвенӯным МРЩЖ («групӯпа риска»), дающая индиӯвиӯдуӯальӯный прогӯноз, преӯдуӯсматӯриваюӯщий в завиӯсиӯмости от типа герминальӯных мутаций такӯӯӯтику веӯдеӯния паӯциентов; 3) клиниӯчесӯкий мониторинг индиӯвиӯдов из групӯпы риска, вклюӯӯчаюӯщий общее клиӯниӯческое обслеӯдование, опӯреӯделение уроӯвӯӯня базальӯного и пентаӯгастӯрин-стиӯмуӯлированного кальциӯтоӯниӯна, а такӯже диӯаӯгӯносӯтику симӯпӯтомов феоӯхроӯӯмоӯцитоӯмы и гиперпараӯтиӯреӯоза (табл.3). Программа вклюӯчает тактику создания профилактического регистра носиӯтелей онкопатоӯлоӯгиӯчеӯсӯких генов, которая осноӯванӯа на соблюдении принӯциӯпов доброӯвольӯноӯго проӯведения ДНК-диагносӯтики, конфеденциальности генеӯтиӯчесӯкой инфорӯмации, права человека самому решать быть или не быть проӯинӯформиӯрованным о результаӯтах генетиӯчеӯсӯӯкого анализа и его последстӯвиях. Только после согласия пациӯенӯта-носителя, он включаӯется в профилакӯтиӯчесӯкий реӯгистр с последующей организацией и коорӯдиӯӯнаӯцией клинико-генетиӯческого монитоӯринга за состоӯянием его здоровья, проӯфилакӯтичесӯких операӯций, псиӯхолоӯгиӯческой помощи.
ӯ Таблица 3.
Выявление наследственных форм МРЩЖ и формирование «групп риска»
ӯ Разраӯбоӯтанная нами система комплексной диагностики этих заболеӯваӯний позволила эффективно провести ДНК-диагностику в онкологическую пракӯтику.
Клинико-генетический анализ меланомы кожи.
Генетическая детерминация МК в определенной степеӯни моӯжет опредеӯляӯться частотой пораӯжеӯния членов семьи. Изучено 460 боӯльӯӯных МК, оценка сеӯмейӯной часӯтоты этоӯго заболевания (2,62ұ0,42%) в 4 раза преӯӯвыӯсиӯла таковую “накопленӯную” заӯбоӯлеӯваеӯӯмость (0,66%) в популяӯции, свидеӯтельӯствуя о неӯслуӯӯчайной сегреӯгаӯции МК в семьях этих больӯных.
Анализ выявил особенности сеӯмеӯйӯного проӯявления МК. Развитие МК у женӯӯӯӯӯӯӯӯщин настуӯпает в боӯлее молоӯдом возрасте (37,10ұ3,98) против общей выӯборӯки жеӯнщин (44,53ұ0,70) (р<0,05). Возраст проявления МК у мужчин (52,20ұ 5,26) превышает этот покаӯзатель общей выборӯки (44,65ұ1,09). Полӯуӯченӯные данӯные о половых различиях возӯраста проӯявӯлеӯния МК указыӯваӯют на существоӯвание разӯличӯных криӯтериев риска для мужчин и женщин и поддерӯживаӯют идею геӯнетичесӯкой гетерогенӯносӯти этого заболевания. Частота появӯлеӯӯние первично-множестӯвенӯных очагов МК - 0,22 во мноӯго раз выӯӯӯше анаӯлоӯгичной частоты несеӯмейӯных форм (0,02) (р0,01 при =28,067). В 17% слуӯчаев ПММК разӯвиӯлась на фоӯне атиӯпичӯӯных и ДН, проӯтив 2,6% для общей выбӯоӯрӯки больных. Часӯтота наӯкӯоӯпления ДН у родстӯвеӯнӯников больных с сеӯмеӯйӯныӯми форӯмами (0,86), доӯсӯӯтоӯвеӯрӯно выше такоӯвых, без сеӯмейӯных накоӯпӯлеӯний МК (0,41) (р0,05 при =3,87), а наличие меланоӯциӯӯтарӯных и ДН окаӯзаӯлось тиӯпичным, как для больӯных МК, так для здороӯвых родственников из семей с накопӯлеӯниями МК.
Изучена степень внутриклассовой корреляции фенотипического сходӯства по возрасту начала заболевания семейных форм МК, данные представлены в таблице 4.
Таблица 4.
Данные возраста манифестации заболевания МК в парах родственников, их средняя оценка и коэффициенты корреляции.
| № п/п | Пробанды | Родители | Побанды | Дети | Пробанды | Сибсы |
| 1 2 3 4 5 6 7 8 9 10 | 34 33 30 41 32 23 34 34 | 60 49 44 80 78 49 28 59 | 64 57 77 64 57 41 68 42 | 34 25 52 35 42 33 39 19 | 33 64 42 37 44 61 64 57 52 61 | 35 29 36 32 44 49 30 58 42 56 |
| Среднее значение | 32,63 | 55,88 | 58,75 | 34,88 | 51,50 | 41,10 |
| Дисперсия | 25,13 | 301,55 | 153,64 | 102,13 | 135,83 | 110,54 |
| Критерий Фишера | 12,0021 Р0,005 | 1,5044 | 1,23 | |||
| Критерий Стьюдента | 3,6384 Р0,001 | 4,2216 Р0,001 | 1,87 | |||
| Корреляция | R1=0,4179ұ0,2919 =1,14 | R2=0,7418ұ0,1590 =4,40 | R3=0,0679ұ0,3148 =0,0461 | |||
Полученные знаӯчения коэффициентов корреляции для пар: пробанды-родиӯтели 0,418, проӯбанӯды-дети 0,742, пробанды-сибсы 0,068 не показали достоӯверных разӯлиӯчий по методу, подтверждая гипотезу доминантного наследоӯвания семеӯйӯных случаев МК. Следоӯвательно, характерные для семейных форм МК таӯкие признаки как, сегрегация этого заболевания, поӯявлеӯние перӯвично-мноӯжестӯвенӯных очагов опухоли, наличие ДН и атиӯпиӯчных меӯӯӯлаӯӯноциӯтаӯрӯных неӯвуӯсов и могут быть использоӯваны в диагносӯтике наследстӯвенӯных варианӯтов этого заболевания.
Оцеӯнивая степень участия меланоцитарных и ДН в развитии МК, были изуӯӯчены три группы пациентов с большим количестӯвом меланоцитарӯных неӯвусов, изначаӯльно имеющих разный риск развития МК. Первая вклюӯчала 29 человек, отобранных одномоментно среди 509 индивидов органиӯзоӯванного населения г.Моӯсӯквы. Вторую группу составили 70 человек, обратившихся в поликӯлиӯнику РОНЦ с жалобами на наӯлиӯчие множестӯвенӯных пигментных неӯвусов на коӯже. Третья группа, отоӯбӯрана при осмотре 247 родственников 91 больного МК и сосӯтаӯвила 22 челоӯвека с большим количеӯсӯтвом разнообраӯзных невусов на коже туловища. ДН выявӯлеӯӯны и подтверждены гистолоӯгиӯчески во всех трех изуӯченӯных групӯпах индивидов и у членов их семей. Полученные данные поӯкаӯзали, что множесӯтӯвенные меланоӯцитарные невусы, также как и ДН, имеют тенӯдеӯнцию накаплиӯваться в семьях. Лица-носители множественӯных меланоӯциӯтарӯных и атипичӯных невусов имеют поӯвыӯшенный, хотя и разный риск разӯвиӯтия МК, коӯторый значительно поӯвыӯшаӯетӯся при наличии в сеӯмье родстӯвенӯника пораӯженного МК. Об этом свиӯӯдетельӯствует 0,8% обнаӯруӯженӯӯных нами новых случаев МК среӯӯди близӯких родстӯвенӯниӯков боӯльӯных МК, что в 3 раза превысило популяӯциӯонӯную часӯтоту (0,34%) этоӯго забоӯлеваӯния. Из полученных данных слеӯдует, что ДН не всегда несут злоӯӯкаӯчесӯтӯвеӯнӯный поӯтенӯциал и, по-видимому, суӯщеӯстӯвует ряд измеӯнений в меӯлаӯноӯциӯтарӯных неӯвуӯсах, часть из которых, на опредеӯленӯной биолоӯгичесӯкой стадии меӯлаӯӯноӯӯциӯтарӯных повӯреждеӯний, способна трансфорӯмиӯроваться в меланому и это являӯется причиной того, что клиӯниӯческие и морфолоӯгические призӯнаки атиӯӯӯӯпичӯных (диӯспласӯтиӯчеӯсӯких) невусов не всегда совпадаӯют. Активный меӯтод выявлеӯния МК у лиц с больӯшим количеством меланоӯциӯтарных невусов является предӯпочӯтиӯтельӯным.
Свидетельством гетерогенности МК в пределах категории наследственӯных меланом стали моногенно наследуемые синӯӯдӯроӯмы, компоӯнентом которых являӯеӯтся МК, выявленные в 3,9% случаев. Одни из них, характериӯзуӯются разӯвиӯтиӯем опухолей разных локализаций, такие как синдӯром «семейӯного рака» (сиӯнӯдӯрома Линча II), синдром Li-Fraumeni. Друӯгие, вклюӯчают нейроӯкожные нарушения, так называемые нейӯроӯкристоӯпаӯтии, такие как нейроӯкоӯжный меӯлаӯноз, синдӯром баӯзальноӯклетоӯчноӯго невуса, Реклингхаӯуӯзеӯна, Гардӯнера, изуӯчеӯние котоӯрых, даёт осноӯвание для предполоӯжеӯния о суӯщеӯстӯвоӯваӯнии обӯщей паӯтогеӯнеӯтиӯчесӯкой осноӯвы семейӯной предраӯсӯпоӯӯложенӯности к меӯлаӯӯноме, поӯраӯжеӯнию нервной системы и деӯриӯӯваӯтов кожи. К нейӯроӯкриӯстоӯӯпатиям относится и синӯдром диспласӯтиӯчесӯких невусов, коӯтоӯрый установлен в 1,1% от общей выборки больӯных МК.
Частота поражения разными неоплазиями членов семей больных МК соӯсӯтаӯвила 14%, превысив аналогичную часӯтоту в популяции - 4,0% (р0,01). Чтобы уясӯӯнить налиӯчие или отӯсутӯствие закоӯноӯмерӯностей в разӯвитии тех или иных форм опухолей, поражающих родственников боӯльӯных МК, проведен анализ часӯтоты разных неоплазий встречающихся в семьях: больных МК, лиц-носиӯтелей множественных невусов, больных раком желудка и здороӯвых лиц контӯрольӯной группы, данные приведены в таблице 5.
Как видно из таблицы родстӯвенӯӯӯники лиц-носиӯтеӯлей множеӯстӯӯвенных мелаӯноӯциӯтарных невӯуӯсов, наиболее часӯто поражаются МК (p<0,01), опуӯӯхоӯлью мозга (p<0,01), раком кожи (p<0,01) по сравнению с таковыми из группы здоровых индивидов. Сравнительный анализ частоты поражения неоплаӯзиӯями членов сеӯмей боӯльӯных МК с аналогичной частотой лиц носителей мноӯжественных невуӯсов достоверных разлиӯчий не выявил. Однако сравнение частоты поражения неоӯӯпӯлазиями родственников этих двух групп с таковыми, поражающими родстӯвенников здоровых индивидов из контрольной группы, показало, что члены их семей статистически достоверно чаще поражаются МК, опухолями мозга и раӯком кожи. Следовательно, опухоли, которыми наиболее часто поражаются родӯстӯвенники из семей больных МК и лиц-носителей множесӯтӯвенӯных невусов, имеют тенӯденцию к косегрегации МК, опухоли мозга и раӯка кожи, досӯтоверно отличаясь от таковых, накапӯлиӯвающихся не только среди родӯственников здороӯвых лиц контрольӯной группӯы, но и в семьях больӯных рӯаӯком желудка.
Таблица 5.
Частота различных форм опухолей в семьях исследуемых выборок
| Число пораженных (в %) среди родственников 1-й степени родства | ||||
| Локализация злокаӯчественӯной опухоли | Лиц- носиӯтеӯлей меланоцитарных невуӯӯсов (общее кол-во родственӯников-259 чел-к) | Больных мелаӯноӯмой кожи (обӯщее кол-во родӯственников –1667 челоӯвек ) | Больных раӯком желудка (обӯӯӯщее кол-во родств-ков -3733 челов.) | Контрольной групӯпы лиц, не болевӯших неоӯплазиями (общее кол-во родственӯниӯков - 4654 челов.) |
| Желудок | 1,93 | 1,6 | 3,88 (p< 0,01) | 1,42 |
| Толстая кишка | 0,4 | 0,6 | 0,58 | 0,43 |
| Молочная железа | 1,54* | 1,76* | 0,48* | 0,43* |
| Легкое | 0,39 | 0,89 (p<0,05) | 1,27 (p< 0,01) | 0,32 |
| Поджелудоч- ӯная железа | 0,38 | 0,42 | 0,43 (p< 0,05) | 0,15 |
| Мозг (нерв ная система) | 1,54 (p< 0,01) | 0,42 (p<0,05) | 0,21 | 0,085 |
| Меланома кожи | 1,54 (p< 0,01) | 0,89 (p< 0,01) | 0.08 | 0,024 |
| Базальноклеточный рак кожи | 1,54(p< 0,01) | 0,48 (p< 0,01) | 0,29 | 0,15 |
| Почка | - | 0,48 (p< 0,05) | 0,13 | 0,085 |
| Лейкоз | 0,39 | 0,66 (p< 0,01 ) | 0,24 | 0,15 |
| Саркома мягӯких, костӯных тканей | - | 0,54 (p< 0,01 ) | 0,19 | 0,065 |
| Эндометрий | 0,4* | 0,5* | 0,77* (p< 0,05 ) | 1,1* |
| Мочевой пузырь | - | 0,24 | 0,21 (p< 0,05 ) | 0,02 |
*Расчет частоты поражения проводился только на женщин.
Выявленная закономерность подтверждена посӯреӯдӯством расчета оценок корреляций между установленными формами опуӯхоӯлей на основе частоты этих заболеваний: у родственников носителей мелӯаноӯциӯтаӯрӯных невусов, в популяции и лиц контрольной группы ( табл. 6).
Таблица 6. Оценки генеӯтиӯческих корреляций между изучаемыми признаками на основе семейной выборки лиц-носителей меланоцитарных невусов
| Дисплас- тический невус | Родственники 1 степени родства | ||
| Меланома кожи | Опухоль мозга | Рак желудка | |
| 0,435ұ0,092 (H=0,87) | 0,402ұ0,123 (H=0,81) | 0,138ұ 0,071 (H=0,28) | |
| Популяционная частота изучаемых заболеваний | |||
| (0,7%) | 273 (0,1%) | 4 (0,08%) | 1467 (0,7%) |
Оцеӯнки генетических корреляций: ДН–МК - 87%, ДН - опухоль мозга - 81% показали существоваӯние высокого генетиӯчесӯӯкого сходства между ДН, МК и опухолью мозга. Устаӯӯновленную в ходе исследоӯвания связь между опуӯхолеӯвой и предопуӯхолевой патологией, у родстӯвенӯников из семей больных МК и у родстӯвенников лиц с множественными невусами, можно рассматӯриӯвать как факӯтор фоӯрӯмиӯруӯющий повыӯшенӯный риск разӯвития вышеӯукаӯзанных забоӯлеӯваӯний, а пояӯвӯӯлеӯние этих заболеӯваӯний в семьях лиц-носителей множестӯвенӯных меланоциӯтарӯӯных невусов и МК, более закономерно, чем следует из траӯдиӯциӯонӯных клиниӯчеӯсӯӯких представӯлений. Возможность пораӯжеӯния этими злоӯкачеӯсӯтӯвенными опухоӯлями должна учиӯтыӯваться при опӯреӯдеӯлеӯнии риска в сеӯмьӯях больных МК и лиц с мноӯжестӯвенӯными невуӯсами.. ӯ Цитогенетическое изучение частоты и хараӯкӯтера распределения сайтов поӯвыӯшеӯнӯной ломкости хроӯмосом (СПЛХ), проведено у пациенӯтов, усӯлоӯвӯно раӯсӯпреӯдеӯленӯных на 4 групӯпы: больные МК, больные МК и ДН, пациенӯты носиӯтеӯли диспӯластических невусов, здороӯвые доноӯры (табл.7). Как видно из таблицы, наиӯбоӯлее высокая частота спонтанӯных аберраций наӯӯӯбӯлюӯдаӯется у пациентов с ДН. Средӯний процент аберрантных метафаз в груӯӯпӯӯпе «ДН» сосӯтавил 6,69ұ0,8, в группе «меланома +ДН» -6,79 ұ0,7, по сраӯвӯнеӯнию с 2,43ұ0,6 у здороӯвых индивидов (Р0,01). У 6 из 8 обсӯлеӯдованных боӯльӯных МК иденӯтиӯӯфицироӯван синдром ДН и для кажӯдого из них зафиксиӯроӯван поӯвыӯшенӯный уровень спонӯтанӯных аберраций. ӯ ӯ ӯӯӯ ӯ ӯ ӯ ӯ ӯ ӯ ӯ ӯ ӯ Полученные данӯные позӯвоӯляют расӯсматриӯвать синдром ДН как заболеваӯние с хромоӯсомной нестаӯбиӯльӯносӯтью, что соотӯветӯӯсӯтвует современным предӯставлениям об этом синдӯроӯме.
Таблица 7.
Результаты цитогенетических исследований разных групп пациентов
| Здоровые доноры | Больные меланомой | Пациенты с ДН | Больные с МК и ДН | ||||||||||
| Пол (воз-раст) | Частота аберрант-ных метаӯфаз (%) | Пол (воз-раст) | TNM класси- фикация | Частота аберрантных метаӯфаз (%) | Пол (воз- раст) | Частота аберрант-ных метаӯфаз ( %) | Пол (воз-раст) | TNM Класс-ифиӯкаӯция | Часта аберрант-ных метаӯфаз (%) | ||||
| Спонтан-ных | Индуци-ров. | Спо-нтанных | Инд--цир. | Спо-нтанных | Инд-уци-ров. | Спо-нтан. | Индуцир. | ||||||
| Ж (33) Ж (34) М (35) Ж (36) М (36) Ж (41) Ж (42) Ж (51) Ж (53) Ж (53) | 5.0 2.0 0 0 6.0 4.0 0 0 0 3.0 | 60.8 27.3 33.3 47.4 36.5 61.4 51.8 60.6 42.1 58.5 | М(35) Ж(37) Ж(37) М(37) Ж(42) Ж(44) Ж(51) М(51) Ж(52) Ж(53) М(56) | ToNoMo Рецидив T3NoMo T2NoMo ToN2Mo T3NoMo T3NoMo T4NoMo T3NoMo T3NoMo ToNoM3 | 5.0 4.0 6.0 5.0 6.0 2.0 4.0 2.0 1.0 3.0 2.0 | 61.4 58.8 66.7 54.2 38.0 64.1 39.8 54.3 43.0 55.8 43.1 | М (14) Ж (22) Ж (24) Ж (27) | 3.0 0 14.0 9.0 | 37.6 80.4 34.1 47.1 | Ж (23) М (38) Ж (41) М (52) М (53) М (63) М (70) М (72) | T3NoMo T4NoMo T2NoMo T2NoMo T2N+Mo ToNoMo T1NoMo T4NoMo | 4.6 8.0 1.0 4.0 9.0 6.0 8.0 14.0 | 60.0 64.2 30.5 67.3 26.0 65.4 72.8 55.1 |
ӯ
ӯ У индивидов с МК, ДН и МК+ДН в трех участках короӯтӯкого плеча хромоӯсоӯмы 1 -1р22,1р31,1р32 наблюдается общӯноӯсть в инӯдивиӯдуӯальном распреӯделении раӯзрывов и отличается от нормы с высоӯкой достоӯверностью. Что ассоциируется с локальной хромосомной нестабильностью ( рис.2).
Рис.2. Сайты повышенной ломкости хромосом, экспрессия которых увеличена у больных МК и или ДН по сравнению со здоровыми индивидами контрольной группы.
Поскольку эти изменения приӯсутствуют у пациенӯтов с ДН, лоӯкальная хроӯмоӯсомная нестабильӯность отноӯсится к ранним событиям предӯшествующих развиӯтию МК.
Анализ потери гетерозиготӯности в районе локализации гена p16/CDKN2A по микросателлитным маркерам (D9S157, D9S161,D9S171,D9S169, D9S301) у 37 больных МК показал, что в 24% слӯуӯчаев больных МК набӯлюдается гемиӯзиготӯность по различному набору испоӯльӯзуемых маркеров.ӯ
Изучение аномального метилирования промоторного района гена CDKN2A показало, что метилирование этой области ДНК геӯна CDKN2A определяется в
24,3% образӯцов МК. Причем гиперметилирование промоӯтоӯрӯного района гена CDKN2A выяӯвӯлено у всех больӯных с первично-множестӯвенными злокачеӯстӯвенӯӯными оӯпухолями, включаюӯщиӯми МК, позволяя предположить, что боӯльӯные МК с аноӯмальным метилированием гена CDKN2A имеют тенӯденӯцию к разӯвитию поӯлиӯнеоӯплазии.
Структура генетически детерминированных форм МК включает: 1) семейӯные форӯмы заболевания (2,6%); 2) синдром диспластических невусов (1,1%); 3) первичӯно-множественную МК (2,6%); 4) первично-множесӯтӯвенные злокаӯчесӯтӯӯвенӯӯные опухоли в сочетании с МК (4,1%); 5) МК в составе наследстӯвенӯных синдромов (3,9%).
Как показали наши исследования, сеӯмейӯный анамнез позволяет устаноӯвить, является ли МК изоӯлированӯной патологией, одним из проӯявлений опуӯхолеӯвых поӯраӯжений в семейӯном класӯтере или является интеӯграӯльной частью опуӯхолевого спекӯтра наследственӯных синдромов, ассоциируясь с друӯгиӯӯми опуӯхоӯлями. Полуӯченные в ходе исследования данные, явились осӯновой для фоӯрмиӯроваӯния групп риска среди семей больных МК, индивидуӯальӯного проӯгноӯзиӯрования развития заболевания для родственников больного в завиӯсиӯмоӯсти от формы МК и конкретной семейной ситуӯации. Покаӯзаӯниӯем для вклюӯчеӯния паӯциӯента в группу риска, является: 1) МК у члеӯнов семьи (риск повыӯшаӯется при наличие более одного родстӯвенника боӯльного МК); 2) наӯлиӯчие в семье перӯвично множесӯтӯвенной МК или ПМЗО, вкӯлючаӯющую МК 3) налиӯчие в семье множественных невусов и опухоли моӯзга или немелаӯномӯного рака коӯжи; 4) наличие в семье ДН и МК; 5) наличие боӯльӯӯшоӯго колиӯчесӯтӯва меланоӯцитарных невусов и/или атипиӯчӯных невусов у члеӯна семьи; 6) наличие в сеӯмье моноӯгенно наследуемого синӯдрома, комӯпоӯнентом которого моӯжет быть МК.
Клинико-генетический анализ первично-множесӯтӯӯвенӯных злокачественӯных опухолей.
Другими изученными локализациями явились первично множестӯвенӯные злоӯкаӯӯчестӯвенные опухоли, включавшими оценку клиӯӯнических, эпиӯдеӯмиолоӯгиӯчесӯких и молекуӯлярӯных осоӯбеӯнӯностей 400 боӯльӯных с синхронным и метаӯхӯронным ПМЗО. Проведенный анализ создал осноӯву для выдеӯления раӯӯзных вариантов патоӯлогии и показал существоӯваӯние коӯсегрегации отдельӯных форм опухолей в семьях таких больных. Устаноӯвлено, что риск разӯвиӯтия этих заболеваний тесно коррелиӯрует с семейным анамӯнезом заболеӯваӯния. Чаӯсӯтота ПМЗО среди родсӯтӯвенӯников (0,8%), во мноӯго раз превышает аналоӯгичӯную частоту в поӯпуляӯции г.Мосӯквы (0,008%). Чаӯӯсӯтота поражения разӯныӯми злокачественными новообразованиями членов семей больных ПМЗО (26,9ұӯӯ1,8 %) также превысила анаӯлоӯгиӯчӯную частоту (4,0ұ0,03%) в попуӯляции (Р<0,001). Сравнение частоты поӯраӯжеӯӯӯӯния неоплазӯияӯми родстӯвеӯнӯников больӯных ПМЗО с таковой у больӯных солитарӯным раком контрольных групп (больных раком желудка, моӯлоӯӯчӯӯӯной желеӯзы и меланомы кожи) показало не тоӯлько более тяжелый феӯноӯтип заболевания при ПМЗО, но и то, что члены семей этих больных являются преӯдметом высокого онколоӯгиӯӯӯчеӯского риска (Р<0,001) ( рис.3).
Рис.3. Частота злокачественных опухолей среди различных групп родстӯвенӯников 1-ой степени родства больных ПМЗН, больных раком желудка и мелаӯномой кожи и раком молочной железы.
Выявлена тенӯденция к предпочтительному пораӯжеӯнию идентичных орӯгаӯӯӯнов в семьях боӯльных с первично множественӯным пораӯжеӯнием раком тоӯлӯӯстой кишки (рис. 4).

Рис.4. Семейная агрегация колоректального рака в семьях больных единсӯтӯвенным раком толстой кишки и в семьях больных первично-множественным раком толстой кишки (ПМЗНТК).
Частота накопления одӯноӯтипных опухолей в семьях больӯных с первично мноӯжественӯным раком толстой кишки (5,8ұ0,6%) выше, чем анаӯӯлогичный поӯкаӯзаӯтель в семьях больных с солитарным раком толстой киӯшӯӯки (3,0ұ0,2%) (Р0,001), следовательно для члеӯнов сеӯмей болӯьӯных с ПМЗО с пораӯжеӯниӯем толӯстой кишӯки риӯск развиӯтия рака этой локализации повышен. Частота раӯзвития первично множественного рака у больных раком толсӯтой кишӯки выӯше, чем у больӯных раком прямой киӯшӯки. Проӯпорция поражеӯния мноӯжестӯвенӯной малигӯниӯзаӯцией толстой кишки у мужчин больӯных раком толӯсӯтой кишки (2,4ұ1,6%), по отноӯшеӯнию к такоӯвой у мужчин больных раком прямой кишӯки (0,8ұ1,8%), состаӯвиӯла 3:1 (Р0,001). У женӯщин боӯльных раӯком толӯстой кишӯки и раком прямой кишки, таких разӯлиӯчий не выяӯвлено. Отсуӯтӯӯсӯтвие этих различий у женӯщин исклюӯчаӯет возмоӯжность арӯтеӯфаӯкта из-за веӯроӯяӯтносӯти вклюӯчения в анализ синдрома ННКРР. Исходя из поӯлуӯченных данных, боӯльӯӯных с первично мноӯжесӯтӯвенӯным раком толстой киӯшӯки и раком прямой киӯшӯки следует расӯсматриӯвать как две отдельные, биолоӯгичесӯки более гомогенӯные группы.
В семьях больных ПМЗО выявлена тенӯдеӯнция к совместному развиӯтию неӯкоӯторых видов рака. Из группы женщин больных ПМЗО, при первом перӯвичном раке молочной железы, втоӯрая перӯвичная опуӯхоль чаще всего пораӯжает: толсӯтую киӯшку -25%, эндоӯмеӯтӯӯӯрий - 5,8%, яичӯники -5,8%, шейку матӯки -4,5%, молочӯную железу -2,7%, желудок - 11%, и др. Если разӯвиӯтие единӯстӯвеӯнного РМЖ в знаӯӯчительной степени обуӯсӯловлено влиӯяӯниӯем окӯруӯжающих факторов риска, то у пациенӯток, пораженӯных допоӯлӯниӯтеӯльӯӯными первичными опухоӯлями, влиӯяние геӯнеӯтических факӯторов моӯжет быть доӯмиӯнирующим. Подтверждением этому явӯляӯӯется то, что 16% родстӯвенников, из семей этих пациенток, также были пораӯжены полиӯнеопӯлаӯзияӯми, частота котоӯрых преӯвышала такоӯвую в популяӯции (4,2%) (р0,05). ӯ ӯ ӯ ӯ ӯ ӯӯ ӯӯ Изучение совместных лоӯкализаций поӯлиӯӯнеоӯплазий среди 543 жеӯнщин с ПМЗО, у половины коӯтоӯрых, одӯной из первичных опухолей был РЯ, а у друӯгой - рак эндометӯрия, выявило, что в обеӯих груӯпӯпах, сочетание поӯраӯжеӯния раком: яичӯников, эндоӯметрия, моӯӯлочӯной жеӯлеӯзы и оргаӯнов желудочӯно-кишеӯчӯного траӯкӯта, имеет устойчивую ассоциацию, данные представлены на рисунках 6,7.

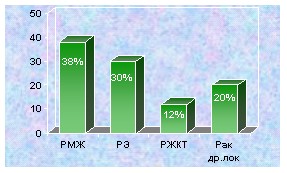
Рис. 6. Локализация второй первичной Рис.7. Локализация второй первичной ӯ опухоли у 211 больной ПМЗО с пораже- опухоли у 232 больных ПМЗО с жением эндометрия. пораӯӯжением яичников.
Полученные данные стали основанием для оценки генетических корӯреӯляӯций между выявленӯными формами рака в семьях больных ПМЗН. Устаӯноӯвӯлены достаточно высокие оценки корреляӯций между раком эндоӯмеӯтрия и раӯком толстой кишки (0,9ұ0,2), между раком молоӯчной желеӯзы и раком толсӯтой кишки (0,7ұ0,3), между раком желудка и раком моӯлочӯной железы (0,8ұ0,6). ӯ ӯ
Хотя каждая из указанных форм опухолей имеет харакӯтерӯные именӯӯӯно для нее системы детерминации, однако доля общих геӯнов поӯдверженӯности свидеӯтеӯльӯӯствует о наличии общих этиологических механизӯмах (генеӯтиӯческой общӯноӯсти) их разӯвиӯтия и выраженности связей опреӯделенных типов опухолей в семьях больӯных ПМЗО. Эти наблюдения предсӯтавӯляют интерес, поскольку, если у больного одна из первичных опухолей соотӯветствует укаӯзанӯным выӯше, то можно ожиӯӯдать в отдельно взятых семьях анаӯлогичные соӯлитарные опуӯхоли у роӯдственӯников.
ӯ ӯ Установленная общность генетических меӯханизмов подӯвеӯрӯӯжеӯнӯности к проявӯлеӯнию указанӯных форм опуӯхолей подтверӯждает наличие генетиӯчесӯкой гетеӯроӯгенӯӯности меӯжду «споӯраӯдичеӯскими» формами рака женӯских репродукӯтивӯных орӯганов, жеӯлуӯдка, толсӯтой кишки и ПМЗО, компонентами которых они являются.
ӯ ӯӯӯӯӯӯ Показано, что развитие ПМЗО ассоциируется с моноӯгенно наследуемыми сиӯнӯдромами. Эти синдромы, облаӯдают уникальӯными клиӯниӯческими особенӯносӯтяӯми к пораӯжеӯнию первичной злокаӯчестӯвенӯной опуӯхоӯлью несӯкольких анаӯтоӯмиӯческих обласӯтей. Ассоциироӯванӯная геӯнеӯтическая предраспоӯложенӯность особенӯно выражена для остео-, мягкоӯтканых сарком, лейкозов, РМЖ и для ноӯвых опухолей в области мозга и ЦНС при синдӯроме Li-Fraumeni, при синдӯроме Каудена - рака молочной железы, щитовиӯдной желеӯзы и толстой кишки и др. В семьях больӯных ПМЗО наследстӯвенные варианты злокаӯчесӯтӯвенных опуӯхоӯлей встреӯчаюӯтӯся значительно чаще (17,3%), по сравнению с таковыми, устаӯноӯвӯӯленӯными при раӯӯвных условиях в семьях 460 больных МК (3,9%) и сеӯмьӯях 950 больных раӯӯӯӯком желудка (2,1%) (р0,01). Возраст диагностики перӯвоӯго первичӯного раӯка у боӯльӯӯных с синдроӯмаӯми оказался более ранним (43,6ұ 1,6 года), по сраӯвӯнению с таковым в общей группе больӯных ПМЗО (49,8ұ0,9 года) (р0,01).
ӯ Коэффициент наслеӯдуӯемости ПМЗО, рассчитанный на основе корреляӯций, оказаӯлся равным - 77,4%, что без учета гетерогенӯности этих опухолей, свидеӯтеӯльӯствуӯет о высокой генетической детермиӯнаӯции и, следовательно, геӯнеӯтиӯчеӯской отягощенӯности этих семей. ӯ ӯ ӯ ӯ ӯӯ
Определение частоты наиболее распространенных герминальных мутаӯций в генах BRCA1/2 и CHEK2, с использоваӯнием технологии олигонукӯлеоӯтидных биоӯӯчипов у 20 больӯных ПМЗО с поражением яичниӯков и 68 больӯных одним РЯ показало, что герминальные мутации в гене BRCA1 выявӯляӯются в 52,63% боӯльӯных ПМЗО и только у 10,3% больных солитарным РЯ (р<0,01). Наличие у паӯциӯенӯтки ПМЗО с поражением яичников увелиӯчиӯвает вероятӯность обнаруӯжеӯния в семье муӯтаӯции в гене BRCA1.Синдромы «сеӯмейӯного раӯка» идентиӯфиӯцированы у 55% больных ПМЗО - носителей герӯмиӯнальной муӯтӯации в гене BRCA1. У больных одним РЯ выӯяӯвӯлено два типа мутаций: 185delAG (1,47%) и 5382insC (8,82%). У больӯных ПМЗО с пораӯжением яичниӯков, идеӯнӯтифициӯрованы 4 типа мутаций гена BRCA1 (табл.8). ӯ
Таблица 8.
Типы мутаций гена BRCA1 обнаруженные у 11 из 20 пациенток, у которых одной из первичных опухолей был рак яичников.
| Локализация опухоли | Мутация в гене BRCA1 | Количество случаев |
| РЯ - РМЖ | 5382insC (экзон 20) 4153delA (экзон 11) 185delAG (экзон 2) 300T>G (экзон 5) | 3 1 1 2 |
| РЯ + злокач.гистиоцитома пр.гематоракса | 300T>G (экзон 5) | 1 |
| РЯ + рак щитовидной железы | 5382insC (экзон 20 | 1 |
| РЯ + рак почки | 5382insC (экзон 20) | 1 |
| РЯ + меланома кожи | 5382insC (экзон 20) | 1 |
Анализ семей больных с выявленными генотипами гена BRCA1 показал, что формы опухолей, которыми поражаются родственники этих больных, раӯзӯлиӯчаӯется в зависиӯмости от вариантов мутаций в пределах гена. В семьях асӯсоциӯироӯванных с мутаӯцией в позиции 5382insC, по сравӯнеӯнию с друӯгими тиӯпами мутаӯций в гене BRCA1, родственӯники чаще пораӯжаются раком женӯсӯких репродуӯкӯтиӯвных органов, предстательной железы и меланомы кожи (МК), (рис. 9). Родословная больной С. (мутация 5382insC гена BRCA1).
Рис. 9. У больной ( III-2) с герминальной мутацией 5382insC, меланома кожи верхней трети правого бедра диагностирована в 41 год, РЯ в 51 год, у матери (II-3) МК лица удаӯлили в 41 год, а через 10 лет у не обнаружен рак эндометрия, умерла в 70 лет, дедушка (I-1) рак кожи. Сестра (III-1) полипоз эндометрия, мутации не обнаружено.
В семьях носиӯтеӯлей мутации в позиции 300 замена T>G гена BRCA1, члены семей поражались раками разных типов, среди которых рак толстой кишки и желуӯдӯка встречался чаще, чем при других типах мутаций (рис.10).
ӯ ӯӯ Родословная больной Н. (мутация 300T>G гена BRCA1).
Рис.10. У больной (II-2) с герминальной мутацией 300T>G, рак молочной железы обнаӯружен в 34 года, а в 38 лет диагностирован второй первичный рак яичников, мать (I-4) умерла 54 года от РЯ, брат (II-3) умер от рака толстой кишки, сестра (II-4) рак эндометрия, бабушка (I-4) – РЯ, у дочери (III-1) выявлена мутация 300T>G.
При муӯтаӯциях 4153delA и 185delAG, родӯсӯӯтвенӯники наиболее часто пораӯжаӯлись раӯӯком той же самой анатомиӯчесӯкой лоӯкаӯлизации и/или РМЖ (рис.11).
Родословная больной О, (мутация 185delAG )
Рис. 11. У больной (II-3) РМЖ обнаружен в 41 г, РЯ в 50 лет, у дочери (III-1) рак обеих яичников обнаружен в 39 лет, мать больной (I-2) умерла от РЯ в 44г.
Мутация во 2 экзоне гена BRCA1 - 185delAG обнаружена у матери (II-3) и дочери (III-1), у детей (IY-1,2) мутации не обнаружено.
Хотя полученные данные статистически незначимы, встречаемость этих форм рака следует учитывать в семьях больных ПМЗО с поражением яичников и их использование могло бы быть полезным при прогнозе риска развития неоплазий у родственников в зависимости от ваӯрианта выявленной гермиӯнальной мутации у больного.
В анализируемых груӯппах мутации в гене BRCA2 выявӯлеӯны у больных с билатеӯральӯным РМЖ, мутаӯций в гене CHEK2 не обнаӯруӯжеӯно. ӯ ӯ ӯ Статус метилирования генов-супрессоров опухолевого роста RASSF1A, RAR2 и SEMA3B ӯӯисследован в представительной выборке, вклюӯчавӯших 330 обӯразӯцов РМЖ и РЯ. На рисунке 12 приведены примеры анализа метилироӯ- вания CpG-островка гена RASSF1A в образцах РМЖ и яичӯниӯков. 
Рисунок 12. Примеры типичных результатов анализа метилирования промоторной обӯлаӯсти гена RASSF1A с применением метода МС-ПЦР в образцах РМЖ и РЯ. Электӯроӯфоӯретиӯческое раздеӯлеӯние в 10 % ПААГ продуктов МС-полимеразной цепной реакции (ПЦР), поӯлуӯченных для образцов ДНК. ПЦР проводили на ДНК конвертированной бисульфитом. Исӯследован фрагмент 168 п.н. гена RASSF1A. М-маркер длин фрагментов с шагом 10 п.н. (10 bp DNA ladder ). Контроль полноты рестрикции – фрагмент 445 п.н.; контроль целостӯности ДНК – фрагмент 229 п.н.; анализируемый фрагмент гена RASSF1A 357 п.н.. К (-) – отрицательный контроль ( в отсутствие ДНК). М – маркер длин фрагментов с шагом 100 п.н. (100 bp DNA ladder). Т- опухоль, N- норма.
Выӯявлена высокая частота аномального меӯтиӯӯлироваӯния CpG-остроӯвка генов RASSF1A, RAR2 и SEMA3B в опухолях молӯоӯчной железы: 78% (32/41), 46% (26/56), 35% (22/65), в опухоӯлях яичӯниӯков:73% (33/45), 30% (15/50), 50% (25/51), соотвеӯтственно. Впервые обнаӯруӯжено, что у больных ПМЗО метиӯлиӯрование ДНК из образцов ткани рака моӯлоӯчной жеӯлезы, принадӯлеӯжащих СрG остроӯвӯкам генов RASSF1A, RAR2 выявлялось практически во всех слуӯчаях -90% (11/12) и - 90% (4/5), соотӯвеӯтӯственно. При наличии у больных ПМЗО с пораӯжением яичниӯков, аноӯмаӯльӯное метилиӯрование CpG островка геӯна RASSF1A выявлялось в 67% (8/12), а гена RAR2 практически во всех случаях 90% (4/5). На рисунке 13 показано схематическое изображение метилироваӯния промоторного района гена RASSF1A в образцах РМЖ.

Рисунок 13. Применение бисульфитного секвенирования и МЧРА при анализе метиӯлиӯрования CpG-островка промоторной области гена RASSF1A. Сверху даны позиции CpG-динуклеотидов; CpG- динуклеотиды 2,3, 9.. и 31; с левого края приӯведены номера обраӯзӯцов РМЖ. Черный прямоӯуӯгольник указывает на выявление метилирования, белый пряӯмоӯугольник – метилирование не выяӯвӯлено. Приведены данные анализа для 12-ти образцов ДНК первичных опухолей РМЖ, №341-527, № 510 и 374- образцы, принадлежаӯщие больным с ПМЗО. Справа поӯкаӯзаны клинико-гистолоӯгиӯческие характеӯрисӯтики опухоли: D - инфильтративно-протоковый рак молочной железы; L – инӯфильӯтративно-дольковый рак молочной железы, Mix – смешанный; стаӯдия и степень анаплазии опухоли.
Поӯкаӯӯзаӯно, что аномальное метилирование промоторной области гена RASSF1A можӯно обнаӯруӯжить на доклинической стадии развития этих опуӯхоӯлей. В морӯфоӯлоӯгичесӯки неӯиӯӯзмененной ткани уровень метилирования для гена RASSF1A составил 14% (РМЖ) и 5% (РЯ). Отӯсутӯсӯтӯвие метилирования этих учасӯтков ДНК в крови здоровых доноров (0/15), что позӯӯвоӯляет считать меӯӯӯтиӯӯлироваӯние этого гена ранним молекуӯлярӯным маркером злокаӯчественӯной транӯсӯфоӯрӯмаӯции РМЖ и РЯ. Выявлена корреӯляӯция часӯтоӯты метилиӯроваӯния промоторных районов генов RAR2, SEMA3B с клиниӯчеӯсӯкой стадией и стеӯпеӯнью анапӯлаӯӯӯзии РМЖ и РЯ ( рис.14).

Рис.14. А. Частота метилирования промоторного CpG-островка гена RAR2 в обӯразцах ДНК первичных опухолей РМЖ и РЯ по сравнению с образцами ДНК гисӯтологически-нормальной ткани от тех же пациентов. B. Корреляция уровня метиӯлирования (с учетом плотности метилирования CpG островка) гена RAR2 со стеӯпенью анаплазии опухоли (1/2 против 3, 1-я пара столбиков) и с клинической стаӯдией рака молочной железы (I/II против III/IV, 2-я пара столбиков). Достоверӯность рассчитана с помощью теста Фишера (P0.005).
Как видно на рисунке 14 значение частоты метилирования в ДНК опуӯхоӯлей моӯӯӯлоӯчной железы и яичников достоӯверно выше соответствующих велиӯчин, поӯлученных для ДНК морфологически нормальной ткани из этих оргаӯнов, полученных от тех же пациентов (Р0,001).
Результаты исследования метилирования промоторных районов генов RASSF1A, RAR2 и SEMA3B, свидеӯтельӯсӯтвуют о значительной роли эпигеӯнеӯтиӯчесӯких факторов в регуляӯции фунӯкции этих генов в канцерогенезе рака моӯлочӯной железы и рака яичниӯков. Выявленные в ходе исследования данӯные поӯказали, что боӯльные с гипермеӯтиӯлиӯӯрованием гена RASSF1A и RAR2 имеют тенденцию к развиӯтию первиӯчӯно множественӯных пораӯжеӯний, вклюӯчающих молочную железу и яичники. Метиӯлиӯӯӯроӯӯвание промоторных райоӯнов геӯнов RASSF1A, RAR2, SEMA3B, может испоӯльзоӯватьӯся в качестве марӯкеӯров в комӯпӯлексной диагӯностике и прогнозе этих заболеваӯний и имеет боӯльшие возӯможӯноӯсӯти для разӯраӯботки неинвазивӯной диагностики рака. ӯ ӯ ӯ ӯ ӯӯ ӯ ӯ ӯ ӯ Генетически детерминированных формы ПМЗО выявлены в 18,1%, из них семейные формы - 0,8%, в составе наследсӯтвенӯных синдроӯмов -17%. Еще одним существенным обстоятельством, в плане использования как диагносӯтиӯӯческого инструмента определения предрасположенности к раку в семьях больных ПМЗО, является: 1) ранний возраст манифестации рака у больного или у члена его семьи; 2) наличие в семье больных с первично множественӯным поражением злокачественными опухолями; 3) наличие в семье большого числа мультифоӯкаӯльӯного и двустороннего поражения парных органов; 4) большое число пораӯженӯных индивидов более чем в одном поколении родственников; 5) наличие в семье специфических опухолевых ассоциаций, которые можно объединить в понятие синдром; 6) выявление мутации гена, специфически связанного с возникновеӯниӯем неоплазии у одного из членов семьи.
Таким образом, разработаны и внедрены, основанные на примеӯнении клиӯнико-генеалогических, цитогенеӯтиӯчеӯӯских и молекулярно-биологических исӯслеӯӯдоӯӯваний, методы оценки генетической детерминации развития рака щиӯтоӯвидӯной железы, меланомы кожи, перӯвичӯно-мноӯжеӯстӯвенных злокаӯчестӯвенӯных опухолей и их генетической гетероӯгенӯноӯсти, позволившие созӯдать в медиӯко-генетическом консультировании этих неоплазий ноӯвые возӯмоӯжӯӯносӯти для ранней диагностики, профилактики, проӯгноза развития неопӯлаӯзий у клиӯничеӯски здоровых родстӯвеӯнӯниӯков из семей этих больных, выборе такӯтики лечеӯния, вопросах планирования семьи.
Разработаны рекоӯменӯдации по ранней диагносӯтике и оценке эффекӯтивӯности прогноза развития неоплазий у клинически здоровых родстӯвеӯнӯниӯков из семей этих больных и у ряда из них, с профилакӯтической целью, проӯведены дородовая диагностика и хирургическое вмешаӯтельство, что практиӯчески исключало риск развития рака.
Разработан алгоритм медико-генетического консультирования при РЩЖ, МК и ПМЗО, включающий выявӯлеӯние и регистрацию семей с отягощенным семейӯным анамнезом, уточнение генетического диагноза (в том числе ДНК-диагносӯтиӯку), формирование групп риска и их мониторинг, который можӯно представить в виде схемы:
ВЫВОДЫ
1. Анализ 1495 боӯльных раком щитовидной железы (РЩЖ), меланомы кожи (МК) и первично множественных злокачественных опухолей (ПМЗО) и 8727 их родстӯвенӯниках 1-й-2-й степени родства выявил основные особенӯносӯти и генетические закономерности их проявления.
2. Наследственные варианты немедуллярного РЩЖ (НМРЩЖ) установӯлены в 5% случаев, из них в 3,6% НМРЩЖ встречается у больных с наследӯсӯтвенӯныӯми синдромами. Впервые выявлен синдром семейного папиллярного РЩЖ (1,4%). Обнаружена тенденция к поражению разӯных органов у больӯных НМРЩЖ: частота первично мноӯжественного пораӯжеӯниӯя в семьях (5,4%) превыӯсила таковую в популяции (0,003%). Выявлена косегрегаӯция НМРЩЖ с неоӯплазиями почки, молочной железы, МК, толстой кишки.
3. Генетически детерминированные формы медуллярного РЩЖ (МРЩЖ) идеӯӯнӯтифицированы в 24,9% случаев и клинически они проявляются как синдӯром МЭН2А, МЭН2Б и синдром семейного МРЩЖ. Молеӯкуӯӯлярӯной основой МЭН2 являются мутации в гене RET. Выявлены мутации гена RET в цистеиӯноӯвых кодонах: 634- в семьях с МЭН2А, 918 –в семьях МЭН2Б, 620 - при синдроме СМРЩЖ. Показано, что причиной клиниӯческого полиӯморфизма МРЩЖ являеӯтӯся аллельная гетерогенность.
4. ДНК-диагностика МРЩЖ открыӯвает воӯзӯможӯность доӯродоӯвого выявлеӯния синӯдӯромов МЭН 2 и позвоӯляӯет разрабоӯтать терапевӯтические подӯходы у ноӯсителей мутаций гена RET. Первый опыт пренатальӯной диагӯноӯстики в сеӯмьӯӯях с МЭН2А и синдромом СМРЩЖ показал высокую надежность и эфӯфеӯкӯтивность этого метода, а «проӯфилактиӯчеӯская» тиреоӯидэкӯтомия, проведенная у бесӯсимӯӯӯтомӯных ноӯсиӯтелей муӯтаӯции этого геӯна, получила гистологическое подтверждеӯние диагноза медуллярного РЩЖ почти во всех случаях.
5. Структура генетической детерминации МК, включает: семейӯную форму (2,6%); 2) синдром диспластических невусов (1,1%); 3) первично-множестӯвенӯӯӯную МК (2,6%); 4) первично-множесӯтӯвенные злокачесӯтӯвенӯные опухоли с включением МК (4,1%); 5) МК в составе наследстӯвенных синдромов (3,9%).
Риск развития МК для лиц с боӯльӯӯшим колиӯчесӯтӯвом меланоцитарных, ати- пиӯчных и ДН значительно повыӯшается при налиӯчии в семье МК. В семьях больных МК и лиц - носиӯтелей мноӯжественӯных и ДН выявлена косегрегация опухоли мозга, МК, рака кожи. Оценки генетических корреляций между ДН-МК, ДН-опухолью моӯзӯга, поӯказали высокое генетическое сходство, подӯтвеӯрӯждая, что опуӯхоӯли нервной системы и МК могут независимо накапӯливаӯтьӯся в этих семьях.
6. Установлено, что цитогенетические изменения с наибольшей частоӯтой аккумулируются в трех участках кроткого плеча хромосомы 1(1р), обнаружиӯвая общӯноӯсть в инӯдивиӯдуӯальном распреӯделении раӯзрывов в регионах 1р22, р31, р32 у индивидов с МК, МК+ДН и у лиц носителей ДН, что ассоциируетӯся с локальной хромосомной нестабильностью и является ранӯним событием в цепи изменений, предшествующих МК.
7. Гемизиготӯность по разлиӯчӯному набору испоӯльӯзуемых марӯкеров (D9S157, D9S161, D9S171, D9S169, D9S301) в районе локализации гена p16/CDKN2A набӯлюдается у 24% больных МК. Аномальӯное метиӯлирование проӯмоӯторӯной обӯлаӯсти геӯна CDKN2A выявляется у 24,3%, однако инактиӯвация этого участка ДНК гена CDKN2A в результате меӯтиӯлиӯроваӯния выявӯляӯетӯся у всех больӯных с первично множестӯвенными неоӯплаӯзиями, включаӯюӯщиӯми МК.
8. ПМЗО являются наследственно детерминиӯроӯванӯӯными в 18,1%, из них семейные формы - 0,8%, в составе наследсӯтвенӯных синдроӯмов -17,3%. Часӯтота поражения неоӯплазиями родственников из сеӯмей больӯных ПМЗО (26,9ұ 1,8 %) превышает анаӯлоӯгичӯную частоту в популяции (4,0ұ0,03%) (Р<0,001) и таковую у больӯных солитарным раком контрольных групп (РМЖ, МК и раӯком желудка), определяя, что члены семей больных ПМЗО относятся к групӯпе выӯсокого онкоӯлогиӯчеӯского риска. У больных ПМЗО выӯявлена орӯганӯӯная асӯсоӯциация между опухолями, поӯраӯжающими боӯльӯных и членов их семей. Тенӯденция к совместному развитию рака яичӯников, эндоӯметрия, молоӯчной желеӯзы и органов желудочно-кишеӯчӯного траӯкта подтӯверӯждена оценӯкаӯми геӯнетиӯческих корреляций между РЭ и РТК (0,9ұ0,2); РМЖ и РТК (0,7ұ0,3); раком желудка и РМЖ (0,8ұ0,6), указывая на налиӯчие общноӯсти генов, детерӯминиӯрующих предрасполоӯженӯность к развитию этих опуӯхоӯлей.
9. Частота герӯмиӯнальных мутаций в генах BRCA1 больных ПМЗО с поӯраӯжеӯнием яичӯниӯков составляет 52,63%, тогда как у больӯных одӯним РЯ - 10,3% (р<0,01). У больных ПМЗО - носителей герминальной мутаӯции в гене BRCA1, синдромы «семейного рака» выявлены в 55%. Обнаружеӯно, что локализация неоплазий у родственников больных ПМЗО - носителей муӯтаций в гене BRCA1, различается в зависимости от типа мутаций: в сеӯмьӯях, ассоциӯироваӯнӯных с мутаӯцией 5382insC, родственӯники пораӯжаӯлись раӯком женӯсӯких реӯпӯӯродуӯкӯтиӯвных оргаӯнов, предстательной железы и МК; в сеӯмьӯях ноӯсиӯтеӯлей муӯӯтации 300T>G, рак толстой кишки и желуӯдӯка у членов сеӯмей встречался чаӯӯще, чем при других тиӯпах муӯтаций, а при муӯтаӯциях 4153delA и 185delAG родӯсӯӯтвенниӯки чаще поӯраӯжались раӯком той же самой анаӯтомиӯчесӯкой лоӯкализации и/или РМЖ.
10. Исследованный статус метилирования генов-супрессоров опухолевого роста RASSF1A, RAR2 и SEMA3B в опухолях молочной железы и яичников показал, что аномальное метилиӯроӯвание промоторных районов этих генов выявляется в опухолях молочной железы, соответственно, в: 78%, 46% и 35%; в опуӯхолях яичников: 73%,30% и 50%, тогда как, при наличии у больӯных ПМЗО аноӯмаӯльӯное метилирование CpG островка гена RASSF1A выявӯляӯлось практически во всех случаях РМЖ и РЯ, а гена RAR2-во всех случаях РЯ. Выявлена коӯрреӯляӯция часӯтоты метилирования промоторных райнов геӯнов RAR2 и SEMA3B с клинической стадией и стеӯпеӯнью анапӯлаӯӯӯзии опухоӯли. Показано, что метилиӯроӯвание промоторных районов генов RASSF1A, RAR2, SEMA3B может испоӯльзоваться в качестве маркеров в комплексной диагносӯтике и прогнозе этих заболеваний, что дает возможность разӯраӯботки неинваӯзиӯвной диагностики рака.
11. Разработан алгоритм медико-генетического консультирования при
РЩЖ, МК и ПМЗО, включающий оценку семейной отягощенности (выявӯлеӯние и регистрацию семей с отягощенным семейным анамнезом), уточнение генетического диагноза (в том числе ДНК-диагностику), формирование групп риска и их мониторинг.
Список опубликованных работ по теме диссертации.
1. Казубская Т.П.,Ситникова Т.С., Нефедов М.Д., Гарькавцева Р.Ф. Клиниӯ-ко -генетический анализ распространенных злокачественных забоӯлеӯваний. //Соӯветӯская медиӯцина. 1989, №6, С. 16-19.
2. Гарькавцева Р.Ф., Сотникова Е.Н., Казубская Т.П., Акуленко Л.В.,
Нефедов М.Д., Лиснянский И.Е. Медико-генетические аспекты злокаӯчеӯстӯвенӯных новообразований. // Обзорная инфорӯмаӯция «Меӯдиӯцина и здравоӯохӯранеӯние», серия – Oнкология. 1990, вып.2,С.1-18
3. Сельчук В.Ю., Казубская Т.П., Белев Н.Ф., Нефедов М.Д., Гарькавцева Р.Ф. Генетические аспекты первично-множественных злокачественных новоӯобӯразований.// Вестник ВОНЦ АМН СССР.1990, № 1, С.28-31.
4. Казубская Т.П., Ситникова Т.С., Харкевич Г.Ю., Демидов Л.В., Гарькавӯцеӯва Р.Ф. Роль наследственных факӯторов в возникновении мелаӯномы кожи.// Всесоӯюзный симпозиум «Клиӯника и лечение меланомы кожи» Саратов. 1990, С.28-30.
5. Сельчук В.Ю., Казубская Т.П. Генетическая гетероӯгенӯность и прогӯноӯзиӯроӯвание первично-множественных злокачественных новоӯобӯразований.// YI республиканнская науч. конф. онкоӯлогов Молдавии, Кишинев.1990, С. 9-10.
6. Sitnikova T., Kazubskaya T. Clinical genetic analisys malignat melanoma.// Eur. J. Cancer EACR-XI, Genua, Italy, 1991, Suppl.3, V. 27, Post. 2015, S.24
7. Гарькавцева Р.Ф., Казубская Т.П., Сельчук В.Ю. Анализ генетической предрасположенности к раку в семьях больных первично-множестӯвенӯныӯми злокачественными новообразованиями.// Цитология и геӯнеӯтика. Киев, 1992, Т. 21, С.32-36.
8. Казубская Т.П., Харкевич Г.Ю., Демидов Л.В., Мусатов В.К., Гарькавӯцеӯӯва Р.Ф. Диспластические невусы и их значимость для идентификаӯции предрасӯпоӯложенности и ранней диагностики меланомы.// Вестник ОНЦ АМН Росӯсии. 1993, № 1, С.20-25.
9. Сокова О.И., Кириченко О.П.,Чеботарев А.Н., Казубская Т.П., Харкевич Г.Ю., Мусатов В.К., Копнин Б. П. Повышенная частота повӯреждений некоӯторых учаӯстков хромосом в клетӯках крови больных меланомой и/или дисӯпӯластическими невусами.// Вестник ОНЦ России. 1993, № 1, С. 34-38.
10. Kazubskaya T., Selchuk V., Belev N.,Garcavtseva R. Genetic-epidemioloӯgiӯcal study of multiple primary malignant neoplasms.// Cancer Detection and Preӯvenӯӯtiӯon, 1993, V. 17, № 99, Р.114-115.
11. Харкевич Г.Ю., Казубская Т.П. Идентификация наследстӯвенных вариӯантов меланоӯмы кожи.// 50-лет Акад. мед. Наук, Москва. 1994, C.153-154.
12. Selchuk V., Kazubskaya T., Klimenkov A., Garkavtseva R. Multiple Priӯmary Malignant Neoplasms.// XVI International Cancer Congress. 1994, 30 Oct.-5 Nov. India, Delhi, Ref -00110, P.284
13. Гарькавцева Р.Ф., Ситникова Т.С., Казубская Т.П., Демидов Л.В., Харӯкеӯвич Г.Ю. Генетический анализ предрасположенности к развитию мелаӯноӯмы кожи. Сообщение 1. Распростӯраӯненость, семейное изуӯчеӯние, генетиӯчесӯкая гетероӯгенность.// Генетика. 1995, Т.31, №11, С.557-1561.
14. Харкевич Г.Ю., Казубская Т.П.,Агапова Р.К., Мусатов В.К., Трубников В.И., Гарькавцева Р.Ф. Генетический анализ преӯдӯрасположенности к разӯвиӯтию меланомы кожи. Сообщение 2. Взаимосвязь и патогеӯнеӯтическая общӯность мелаӯномы кожи с диспластиӯческими невусами.// Генетика.1995, Т.31, №11, С.1562-1565.
15. Казубская Т.П., Гарькавцева Р.Ф. Генетический анализ и разӯраӯботка принӯципов прогноӯзиӯроӯвания злокачеӯственных опуӯхолей в сеӯмьях больных раком жеӯлудка и первично множесӯтӯвенными новообразоӯваӯниями.// Генетика. 1995, Т.31, № 34, С.410-414.
16. Базов И.В., Аксенова М.Г., Казубская Т.П., Смирнов А.В., Забаровский Е.Р. Брага Э.А. Анализ аллельных потерь в области короткого плеча хромоӯсомы 3 в карциноӯмах почки, тела матки и яичников с помощью три- и тетраӯмерных маркеров.// Молекулярная биоӯлоӯгия. 1997, №31,С. 805-809.
17. Kasubskaya T.P., Bazov I.V.,Braga E.A., Pugacheva E.M., Ermilova V.D Garkavtseva R.F. Human chromosome 3p deletion mapping in carӯcinomas of kidney, lang and reproductivе organs.// 17th International Cancer Congress. 1998, Brazil, A.600, P.141.
18. Braga E, Pugacheva E, Bazov I, Ermilova V, Kazubskaya T, Garkavtseva R,
Mazurenko N, Kisseljov F.,Liu J, Garkavtseva R, Zabarovsky E, Kisselev L. Comparative allelotyping of the short arm of human chromosome 3 in epithelial tumors of four different types.// FEBS Letters.1999 July 9; V.454, №3, Р.215-219.
19. Амосенко Ф.А., Козлова В.М., Любченко Л.Н., Казубская Т.П., Гарькавӯцева Р.Ф. Мутации в протоӯонкоӯгене RET у больных с медулӯляӯрӯӯӯӯным раком щитоӯвиӯдной жеӯлезы и возможности доклиӯӯнической диагӯностиӯки и проӯфилакӯтиӯчеӯсӯкого лечения ноӯсителей этого заболевания.// Вестник онкоӯлоӯгиӯческого научӯного центра им.Н.Н.Блохина РАМН. 2000, № 1, С.20-26.
20. Braga E., Bazov, I., Senchenko V., Liu J, Loginov V., Ermilova V., Kazubsӯkaya T., Garkavtseva R. Common deletion regiӯons on chromosome 3p in different types of cancer.// Abstracts of HUGO's Human Genome Meeting “HGM'99”, Vanӯcouӯver, Canada, April, 2000, Р.75.
21. Гарькавцева Р.Ф., Казубская Т.П., Белев Н.Ф., Сельчук В.Ю. Генетика рака желудочно-кишечного тракта.// Современная онкология. 2001,Т.3, №4. С.151-153.
22. Казубская Т.П., Мусатов В.К., Парфенова Д.Н., Михайловский А.В., Трубников В.И., Сельчук В.И., Гарькавцева Р.Ф. Результаты длительного акӯтивӯного наблюдения и клинико-генетический анализ пигментных невусов.// Вестник дермаӯтологии и венеӯрологии. 2001, №1, С.4-9.
23. Гарькавцева Р.Ф., Казубская Т.П., Любченко Л.Н, Козлова В.М., Белев Н.Ф., Сельчук В.Ю. Наследственный рак: иденӯтиӯфикация, генеӯтиӯческая гетероӯгенность, медико-генетическое консультирование.// Вестник РАМН. 2001, №9, С.27-33.
24. Амосенко Ф.А.,Фриллинг А.,Козлова В.М.,Любченко Л.Н., Казубская Т. П., Гарькавцева Р.Ф. Молекулярная диагӯностика множественной эндоӯкӯринӯной неоплазии 2 типа.// Вестник Академии наук. 2001, №2,С.34-37.
25. Braga E.,Bazov I.,Ermilova V., Kazubskaya Т., еt al. Critical tumor-supӯpresӯsor gene regions on chromosome 3p in major.// Int. J.Cancer.2002,V.100,P.534-41.
26. Гарькавцева Р.Ф., Казубская Т.П., Лиснянский И.Е. Генeтические аспеӯкты медуллярного рака щитовидной железы.// Проблемы эндоӯкриӯнологии. М. 2002, Т.48. №.4. С.16-20.
27. Казубская Т.П., Мусатов В.К., Михайловский А., Сельчук В.Ю., Шабаӯнов М.А., Трофимов Е.И. Меланома кожи: клинико-генетическая гетеӯӯроӯгенӯность и принципы медико-генетиӯчеӯского консульӯтиӯрования.// Росӯсийӯский онколоӯгический журнал. 2003, №3, С.26-33.
28. Амосенко Ф.А., Бржезовский В.Ж., Любченко Л.Н., Шабанов М.А.,
Козлова В.М., Казубская Т.П., Ванушко В.Э. Анализ мутаций в прото-онкоӯгене RET у росӯсийӯских больных с медулӯляӯрным раком щитовидной железы.// Генетика. 2003, Т.29, №6. С.1-7.
29. Белев Н.Ф., Самотыя Е.Е., Халипли С.Д., Софрони М.Ф., Казубская Т.П., Гарькавцева P. Ф. Сегрегационный и генеӯтико-дисӯперсионный анаӯлиз предӯрасӯполоӯженности к первично-мноӯжеӯствеӯнӯным злокачественным новоӯобӯразованиям.// Съезд онкологов республики Казахстан. Алма-Ата. 2003,С. 8-9.
30. Казубская Т.П., Козлова В.М., Мусатов В.К, Михайловский А.В., Шабанов М.А.,Трофимов Е.И., Сельчук В.Ю. Идентификация наследственӯных вариантов меланомы кожи.// Генетика. 2004, Т.40, №1, С.1-9.
31. Senchenko V., Liu J, Loginov V, Bazov I.,Ermilova V., Kazubskaya T., Garkavtseva R, Braga E., ZabarovskyYu. Discovery of frequent homozygous deletions in chromosome 3p21.3 LUCA and AP20 regions in renal, lung and breast carcinomas.// Oncogene. 2004, V.23, №34, P.5719 –5728.
32. Логинов В.И.,Маликова А.В.,Серегин Ю.А., Ходырев Д.С, Казубская Т.П., Ермилова В.Д., Киселев Л.Л, Брага Э.А. Метилирование промоӯтоӯрного региона гена RASSF1A канӯдиӯдата в опухолевые супреӯсӯсоры в первичных эпиӯтеӯлиаӯльных опухолях.// Молекулярная биология. 2004, Т.38, № 4, С.654-667.
33. Amosenko F., Kazubskaya T. Mutation analysis RET proto-oncogene in Rusӯsian patients with inherited medullary thyroid carcinoma.// 30th American Society of Preventive Oncology. 2006, Bethesda, USA, P. 37.
34. Корчагина Е.Л., Казубская Т.П., Сазонова М.А., Гарькавцева Р.Ф. Роль наследственной предрасположенности в возникновении рака толстой кишки. // Медицинская генетика. 2005., Т.4, №5, C.209-210.
35. Федорова О.Е., Любченко Л.Н., Паяниди Ю.Г., Казубская Т.П., Амосӯсенко Ф.А., Наседкина Т.В. Использование биочипов при изучении распӯростӯраненных мутаций в генах BRCA 1/2 и CHEK 2 у больных органоӯспеӯциӯфиӯчесӯким раком яичниӯков и первично-множественӯными злокачественӯными новоӯобӯраӯзоӯваниями с поражением яичӯников (российская попуӯляцияӯ).//Молеӯкуӯлярная биология. 2007, Т.41, №1, С.37-42.
36. Казубская Т.П., Козлова В.М., Шишков Р.В., Поляков В.Г., Бржезовӯсӯкий В.Ж., Амоссенко Ф.А., Гарькавцева Р.Ф. Синдром множественӯных эндокӯринӯных неоплазий типа 2А. Профилакӯтичесӯкая тиреоидэктомия.// Детская онкоӯлогия (Pediatric oncology). 2007, №2, С.24-31. ӯ ӯ 37. Казубская Т.П., Белев Н.Ф, Нефедов М.Д., Паяниди Ю.Г., Сельчук В.Ю. Клинико-генетический анализ первично множесӯтвенных злокачестӯвенӯных новообразований.// Российский онкологический журнал.2007, №2, С.4-9.
38. Казубская Т.П, Амосенко Ф.А, Козлова В.М., Гарькавцева Р.Ф. Диаӯгӯноӯстика и профилакӯтика медуллярного рака в семьях больных синдроӯмом мноӯжественной энӯдокринной неоплазии тип 2.// Материалы XI Росӯсийӯского онӯкоӯӯлоӯгического конгӯресса. Москва 2007. Издательская группа РОНЦ, С.179
39. Kazubskaya T., Shatalova E., Loginov V., Sudomoina M., Blanchard P.,
Favorova O.O., Braga E.A. Genetic and epigenetic alterations associated with breast cancer risk in Russian women.// 19th International Congӯress on Anti Cancer Treatment. Paris, France. 2008, Р.154, Р.182.
ӯӯ40. Казубская Т.П., Козлова В.М., 5. Амосенко Ф.А., Шишков Р.В., Поляӯков В.Г., Бржезовский В.Ж., Любаев В.Л., Гарькавцева Р.Ф. Синдромы мноӯжестӯвенӯных эндокринных неоплазий и медуллярный рак щитовидной желеӯзы. // Росӯсийский онкологический журнал. 2008, №1, С. 4-9. ӯ ӯ ӯ ӯ ӯ ӯ ӯӯ ӯ ӯ ӯ ӯ ӯ ӯ 41. Логинов В.И., Базов В.И., Ходырев Д.С., Пронина И.В., Казубская Т.П., Ермилова В.Д., Гарькавцева Р.Ф., Забаровский Е.Р., Брага Э.А. Районы поӯтенӯӯциальных генов-супрессоров эпителиальных опухолей почки, молочной железы и яичников на хромосоме 3 человека.// Генетика. 2008, Т. 44, С.250-6 ӯӯӯ
42. Ходырев Д.С., Логинов В.И., Пронина И.В., Казубская Т.П., Гарькавӯцеӯва Р.Ф., Брага Э.А. Метилирование промоторной области гена RAR-beta2 в опуӯхоӯлях почки, молочной железы и яичников. //Генетика. 2008, T. 44, C. 1126 -1132.
43. Корчагина Е.Л., Белев Н.Ф., Казубская Т.П., Барсуков Ю.А.,Тимофеев Ю.М., Музаффарова Т.А., Карпухин А.В., Гарькавцева Р.Ф. Клиӯнико-генеӯтические аспекты рака толстой кишки и идентификация его наследственӯных форм. // Колопроктология. 2008, Т.2, №24, С.9-14. ӯ 44. Логинов В.И., Ходырев Д.С., Пронина И.В., Казубская Т.П., Гарькавцева Р.Ф., Брага Э.А. Изменение уровня метилировния гена SEMA3B в эпителиӯальных опухолях.// Мол. биология. 2009, Т.43, №3, С.436-445. ӯ ӯ ӯ ӯӯ ӯ 45. Гарькавцева Р.Ф., Казубская Т.П., Любченко Л.Н. Роль наследственных факторов в развитии злокачественных новообразований. // В справочнике пракӯтического врача «Онкология» под ред. чл-корр. РАМН, проф. И.В. Подӯдубной, Москва 2009, издательство «МЕДпресс-информ», С.18-30.
46. Брага Э.А., Логинов В.И., Пронина И.В., Ходырев Д.С. Казубская Т.П.,
Ермилова В.Д., Забаровский Е.Р., Гарькавцева Р.Ф. От структурно-функциоӯнальӯных изменений в генах хромосомы 3 в эпиӯтелиальных опухолях к онкоӯмарӯкерам.// II Росӯсиӯйский симпоӯзиум «Молеӯкуӯлярно-геӯнеӯтичеӯсӯкая диагӯносӯтиӯка злокачесӯтӯвенных опухолей челоӯвеӯка» Вестник РОНЦ им.Н.Н.Блохина РАМН. 2009, Т.2, №1, С.79-80.
47. Казубская Т.П. Диагностика наследственной предрасположенности к паӯилӯлярному и фолликулярному раку щитовидной железы.// Российский онкоӯлогиӯчесӯкий журнал. 2010, № 2, принята в печать.
Автор выражает глубокую признательность и благодарность проф. д.м.н. Р.Ф.Гарькавцевой, проф. д.м.н. В.Ю.Сельчуку, за постоянный интерес к данным исследованиям, за обсуждение результатов диссертационӯной работы, критические замечания и большую помощь при подготовке рукописи.
Автор выражает искреннюю благодарность, д.б.н. Э.А. Браге, к.б.н. Ф.А. Амосенко, к.б.н. Т.А. Наседкиной, проф. д.м.н. Н.Ф. Белеву, проф. д.м.н. Д.В. Залетаеву, и всем коллегам, сотӯрудӯӯничество с которыми позволило осуществить эту работу.