

Роль трансформирующего ростового фактора (tgf ) в прогрессии гепатокарцином
На правах рукописи
МАКАРОВА МАРИЯ ВИКТОРОВНА
РОЛЬ ТРАНСФОРМИРУЮЩЕГО РОСТОВОГО ФАКТОРА (TGF) В ПРОГРЕССИИ ГЕПАТОКАРЦИНОМ
14.00.14 – онкология
АВТОРЕФЕРАТ
диссертации на соискание ученой степени
кандидата биологических наук
МОСКВА
2009
Работа выполнена в НИИ Канцерогенеза Учреждения Российской академии медицинских наук Российский онкологический центр имени Н.Н. Блохина РАМН
Научный руководитель:
Доктор биологических наук Н.Л. Лазаревич
Официальные оппоненты:
Доктор биологических наук, профессор П.М. Чумаков
Доктор биологических наук Н.А. Глушанкова
Ведущая организация: Научно-исследовательский институт физико-химической биологии им. А.Н.Белозерского МГУ
Защита состоится «______» _____________2009 г. в _______ часов на заседании диссертационного ученого совета Д.001.017.01 РОНЦ им. Н.Н.Блохина РАМН по адресу 115478, Москва, Каширское шоссе, 24.
С диссертацией можно ознакомиться в библиотеке РОНЦ им. Н.Н.Блохина РАМН.
Автореферат разослан «______» _________________2009 г.
Ученый секретарь диссертационного совета,
доктор медицинских наук, профессор Ю.В. Шишкин
ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ
Актуальность проблемы
Гепатокарцинома (ГК) является пятой по распространенности и третьей по уровню смертности опухолью в мире [Parkin et al., 2005]. Интенсивные эпидемиологические исследования последних лет позволили идентифицировать множество факторов риска возникновения ГК. Однако ключевые молекулярные механизмы, лежащие в основе гепатоканцерогенеза, остаются недостаточно изученными. В ходе прогрессии ГК происходят изменения активности внутриклеточных сигнальных каскадов и нарушения программы экспрессии гепатоспецифических генов. Одними из основных молекулярных изменений сигнальных путей, идентифицированных для ГК человека, являются нарушения активности Wnt/-катенин сигнального пути, р53, MAPK, JAK/STAT, Ras и TGF сигнального каскада [Aravalli et al., 2008].
TGF является плейотропным цитокином, контролирующим различные свойства клеток организма, такие как пролиферация, подвижность, адгезия и дифференцировка. Генетические изменения компонентов TGF сигнального каскада описаны для опухолей различного происхождения, но его роль в канцерогенезе противоречива, так как TGF в зависимости от клеточного контекста может проявлять либо свойства опухолевого супрессора, либо онкогенные черты. Считается, что в нормальных клетках и на ранних этапах развития эпителиальных опухолей TGF подавляет пролиферацию и индуцирует апоптоз, однако на более поздних стадиях прогрессии он индуцирует эпителиально-мезенхимальный переход (ЭМП), усиливает инвазию и метастазирование [Pardali et al., 2007].
Роль TGF в возникновении и развитии ГК также неоднозначна и недостаточно изучена. TGF1 является значимым ингибитором пролиферации нормальных гепатоцитов. При повреждении печени TGF1 сигнальный каскад активируется, останавливая рост регенерирующих гепатоцитов [Bissell et al., 1995]. С другой стороны, у пациентов с ГК уровень TGF1 в плазме крови и моче повышен [Tsai et al., 1997; Dong et al., 2008]. Иммуногистохимическими методами было показано, что TGF1 продуцируется в более чем 40% исследованных образцов ГК [Abou-Shady et al., 1999].
У млекопитающих выявлены три белка семейства TGF (1, 2 и 3), которые высоко гомологичны по аминокислотной последовательности. Однако при инактивации генов TGF наблюдаются непохожие эффекты, что, по-видимому, свидетельствует о функциональном различии изоформ TGF [Massague, 1998]. Большинство исследований, проводимых на гепатоцитах, сосредоточено на изучении роли TGF1, однако наши результаты свидетельствуют о том, что при прогрессии гепатокарцином может усиливаться транскрипция гена TGF2. Роль такой активации в гепатоканцерогенезе в настоящее время не изучена.
Цель и задачи исследования
Основной целью настоящей работы являлось изучение роли TGF2 в регуляции экспрессии генов, кодирующих ГЯФ и маркеры дифференцировки, а также исследование влияния TGF2 на процессы, ассоциированные с прогрессией ГК. Для достижения поставленной цели были поставлены следующие экспериментальные задачи:
- Проанализировать изменения экспрессии гепатоспецифических генов и генов, продукты которых ассоциированы с прогрессией ГК, в культуре дифференцированной гепатомы человека HepG2 при обработке цитокинами TGF1 и TGF2.
- Провести трансдукцию линии гепатомы HepG2 лентивирусной конструкцией, экспрессирующей TGF2. Исследовать влияние гиперэкспрессии гена TGF2 на кинетику пролиферации, клоногенную активность, уровни экспрессии гепатоспецифических генов; генов, ассоциированных с ЭМП и TGF2-респонсивных мишеней.
- Исследовать вклад латентной и активной формы TGF2 в регуляцию процессов, связанных с активацией TGF сигнального пути. Провести трансдукцию лентивирусной конструкцией, экспрессирующей мутантную форму TGF2, не подвергающуюся протеолитической деградации, и сравнить эффекты от аналогичной конструкции с нормальным вариантом TGF2.
- Разработать систему для подавления синтеза TGF2 в культуре Н33, полученной из низкодифференцированной быстрорастущей ГК мыши. Изучить изменения в кинетике пролиферации, способности к инвазии и профиле экспрессии гепатоспецифических генов в клетках Н33 с подавленной продукцией TGF2.
Научная новизна и практическая
значимость исследования
В представленной работе впервые проанализированы изменения экспрессии генов в дифференцированной гепатоме человека HepG2 при воздействии рекомбинантных цитокинов TGF1 и TGF2. Обработка цитокинами привела к повышению экспрессии генов 3 субъединицы интегрина, остеопонтина, фибронектина 1 и ig-h3 (TGF индуцируемого гена-h3), активация которых ассоциирована с развитием злокачественного фенотипа ГК. Кроме того, было показано, что действие обоих цитокинов приводит к снижению экспрессии генов, кодирующих ГЯФ, которые необходимы для поддержания дифференцировки гепатоцитов.
Мы показали, что экзогенная экспрессия TGF2 в культуре HepG2 вызывает снижение экспрессии ключевых регуляторов гепатоцитарной дифференцировки C/EBP и HNF4, а также к повышению скорости пролиферации и клоногенной активности этих клеток.
Впервые установлено, что подавление продукции TGF2 в культуре клеток низкодифференцированной гепатомы Н33 индуцирует реэкспрессию эндогенных HNF4 и C/EBP. Кроме того, подавление продукции TGF2 привело к снижению подвижности и скорости пролиферации этих клеток.
Полученные результаты свидетельствуют о том, что TGF2 регулирует экспрессию генов, продукты которых контролируют дифференцировку гепатоцитов, и влияет на такие важные свойства клеток, как пролиферация и подвижность. Эти данные свидетельствуют о том, что гиперэкспрессия TGF2, наблюдаемая в дедифференцированных ГК мыши и человека, способствует развитию злокачественного фенотипа опухолевых клеток. Кроме того, полученные результаты демонстрируют возможность регуляции сети ГЯФ и дифференцировки клеток гепатоцитарного происхождения за счет изменения активности нетканеспецифических сигнальных путей. Эти факты способствуют расширению представлений о фундаментальных основах опухолевого роста и прогрессии злокачественных новообразований. Возможность восстановления дифференцированного фенотипа гепатоцитов при ингибировании TGF сигнального пути представляется перспективным подходом для разработки новых методов прогноза и терапии ГК. Анализ TGF-индуцируемых внутриклеточных механизмов будет способствовать развитию методов эффективного подавления продукции TGF для терапии эпителиальных опухолей различного происхождения.
Апробация работы
Диссертация апробирована 16 февраля 2009 г на совместной научной конференции лабораторий механизмов прогрессии эпителиальных опухолей, цитогенетики, регуляции клеточных и вирусных онкогенов и механизмов канцерогенеза НИИ канцерогенеза РОНЦ им Н.Н. Блохина РАМН. Материалы работы докладывались на международном конгрессе «Исследования заболеваний печени» Европейской ассоциации по исследованию печени (Копенгаген, 2009), международной конференции «Ядерные рецепторы и заболевания печени» (Вена, 2009), конференции «Молекулярные механизмы сигнальной трансдукции и рака» Европейской Федерации Биохимических обществ (FEBS) (Спецес, 2007), на 13 съезде Европейской Конференции по раковым исследованиям (ЕССО) (Париж, 2005), и на конференции «Клеточная сигнализация» Европейской Федерации Биохимических обществ (FEBS) (Дубровник, 2006).
Публикации
По теме диссертации опубликовано 12 печатных работ, в том числе 2 статьи в отечественных журналах и 10 тезисов международных конференций.
Структура и объем диссертации
Диссертация изложена на 137 страницах машинописного текста (12 шрифт, полуторный интервал), содержит 30 рисунков, 1 таблицу. Состоит из глав: «Введение», «Обзор литературы», «Материалы и методы», «Результаты исследования и их обсуждение», «Заключение», «Выводы», «Список литературы». Список литературы содержит 231 источник, в том числе 9 в отечественных рецензируемых изданиях.
ОСНОВНОЕ СОДЕРЖАНИЕ РАБОТЫ
Ранее в нашей лаборатории была получена и охарактеризована модель одноступенчатой прогрессии ГК мыши. Модельная система состоит из медленнорастущей высокодифференцированной гепатокарциномы (мГК), выщепившейся из нее in vivo быстрорастущей низкодифференцированной гепатокарциномы (бГК), и полученной из бГК клеточной линии Н33 [Lazarevich et al., 2004]. Ранее методом гибридизации с кДНК микрочипами были проанализированы спектры экспрессии различных генов в мГК, бГК и Н33 по сравнению с нормальной печенью взрослой мыши. Было показано, что существенная часть генов, активировавшихся в ходе прогрессии от мГК к бГК, оказались TGF-индуцируемыми, что указывает на вовлеченность этого сигнального пути в прогрессию опухолей печени [Лазаревич, 2004].
При проверке этого предположения методом ОТ-ПЦР было показано, что в ряду нормальная печень, мГК и бГК наблюдается активация экспрессии различных TGF-респонсивных генов. Важно отметить, что в ходе прогрессии ГК была обнаружена значительная индукция транскрипции гена TGF2. Уровень экспрессии гена TGF1 во всех опухолях превышал детектируемый в нормальной печени, в бГК и Н33 по сравнению с мГК он практически не изменился (Рис. 1).
Кроме того, параллельно с активацией экспрессии гена TGF2, в бГК была снижена транскрипция гена HNF4, который играет ключевую роль в дифференцировке нормальных гепатоцитов в процессе эмбриогенеза и в поддержании гомеостаза нормальной печени во взрослом организме [Лазаревич и Флейшман, 2008].

Рисунок 1. Изменение экспрессии генов HNF4, TGF1 и TGF2 при одноступенчатой прогрессии ГК. ОТ-ПЦР анализ уровней экспрессии мРНК в мГК, бГК, нормальной печени (П), культуре Н33. Для контроля равенства количества РНК, взятой в реакцию, приведены результаты ОТ-ПЦР с праймерами к мРНК глицеральдегид 3-фосфат дегидрогеназы (GAPDH).
Целью настоящей работы было исследование возможной роли гиперэкспрессии TGF2 в прогрессии гепатокарцином. Для этого были использованы две модельные системы: линия дифференцированной гепатомы человека HepG2 и культура быстрорастущей дедифференцированной ГК мыши Н33, которая описана выше. На первом этапе работы было исследовано влияние индуцируемых TGF2 сигнальных путей на экспрессию генов и биологические свойства клеток HepG2. В качестве системы, в которой уровень TGF2, наоборот, повышен, мы использовали культуру ГК мыши Н33, в которой с помощью малых интерферирующих РНК была подавлена продукция TGF2.
1. Изучение TGF -индуцируемых эффектов в клеточной линии HерG2
1.1. Анализ экспрессии генов, ассоциированных с прогрессией ГК, и генов, ответственных за поддержание дифференцированного статуса гепатоцитов, при обработке клеточной линии HepG2 цитокинами TGF1 и TGF2
Для исследования возможного влияния TGF на изменение спектра экспрессии генов, определяющих дифференцировку клеток гепатоцитарного происхождения, а также генов, изменение уровней транскрипции которых ассоциировано с прогрессией ГК, клетки HepG2 обрабатывали TGF1 и TGF2 в концентрации 5 нг/мл в течение 24 или 48 часов. Методом ОТ-ПЦР был проведен анализ экспрессии генов и показано, что действие TGF1 и TGF2 вызывает усиление экспрессии генов 3 интегрина, фибронектина 1, остеопонтина и ig-h3 (Рис. 2). Выбор исследуемых генов был основан на результатах анализа изменения транскрипционной программы при одноступенчатой прогрессии ГК мыши методом гибридизации с кДНК микрочипами. Выбранные гены, во-первых, по литературным данным входили в группу TGF-индуцируемых, во-вторых, их активация была ассоциирована с одноступенчатой прогрессией ГК мыши.
Экспрессия генов 3 интегрина, фибронектина 1, остеопонтина и ig-h3 была повышена по сравнению с контролем при обработке как TGF1, так и TGF2 (Рис. 2). Более того, для гена фибронектина 1 была показана экспрессия сплайс-формы, характерной для трансформированных и эмбриональных гепатоцитов, содержащей EDA домен. Праймеры для ОТ-ПЦР были подобраны таким образом, что при электрофорезе продуктов ПЦР выявлялось 2 полосы: нижняя из них соответствовала взрослой изоформе фибронектина (565 пар нуклеотидов - п. н.), лишенной EDA домена, а наличие верхней полосы (720 п. н.) отображало присутствие в исходной пробе мРНК эмбрионального варианта фибронектина (Рис. 2).


Рисунок 2. Изменения экспрессии TGF-зависимых генов, а также генов кодирующих транскрипционные факторы и гепатоспецифические гены при обработке клеточной линии HepG2 TGF1 или TGF2. ОТ-ПЦР анализ уровней транскрипции генов, ассоциированных с прогрессией ГК. Дорожки 1, 3 – обработка TGF1 24 и 48 ч соответственно, дорожки 2, 4 – обработка TGF2 24 ч и 48 ч соответственно, дорожка 5 – контроль - HepG2 без обработки цитокинами. Для контроля равенства количества РНК, взятой в реакцию, приведены результаты ОТ-ПЦР с праймерами к GAPDH.
Повышенная экспрессия остеопонтина ассоциирована, по мнению ряда авторов, с развитием метастатического потенциала гепатокарцином, также как и экспрессия ig-h3 [Gotoh et al., 2002; Tang et al., 2007]. Ранее было показано, что экспрессия 3 субъединицы интегрина характерна для незрелых и трансформированных гепатоцитов, и опосредует способность ГК к инвазии [Giannelli et al., 2002].
Необходимо также отметить, что действие TGF1 или TGF2 активирует эндогенную экспрессию соответствующих генов, что указывает как на авторегуляцию, так и на взаимную регуляцию их транскрипции (Рис. 2).
Исследования, проведенные на модели одноступенчатой прогрессии, показали, что дедифференцировка является ключевым процессом, сопровождающим малигнизацию ГК [Lazarevich et al., 2004]. Дифференцированный фенотип гепатоцитов определяется экспрессией специфических функциональных генов, транскрипция которых в значительной степени регулируется различными ГЯФ.
Принимая во внимание эти факты, логично было предположить, что воздействие TGF влияет на экспрессию по крайне мере некоторых компонентов регуляторной сети ГЯФ и дифференцировочных маркеров. При анализе уровней транскрипции всего спектра ГЯФ, экспрессирующихся в дифференцированных гепатоцитах, методом ОТ-ПЦР, мы установили, что инкубация HepG2 с TGF1 и TGF2 вызывает подавление транскрипции генов изоформы HNF4, характерной для дифференцированных гепатоцитов (HNF4Р1), C/EBP, HNF1, OC2, FoxA2, а также основного маркера дифференцированных гепатоцитов сывороточного альбумина (Рис. 2).
Необходимо отметить, что при обработке HepG2 цитокинами TGF1 и TGF2 произошло координированное изменение активности двух альтернативных промоторов гена HNF4: активация экспрессии “эмбриональных” изоформ HNF4Р2 (характерных для незрелых и трансформированных гепатоцитов) и подавление уровня экспрессии “взрослых” изоформ HNF4Р1, которые экспрессируются во взрослой печени.
1.2. Изменение экспрессии изоформ HNF4P1 и HNF4P2 при ингибировании передачи сигнала от рецептора TGF
Действие TGF в основном опосредуется активацией рецептора TGF I типа и рецептора II типа. Фосфорилирование внутриклеточных субстратов происходит в результате действия рецептора I типа. Для того чтобы удостовериться, что эффекты, которые оказывает TGF2 на экспрессию гена HNF4, специфичны и связаны именно с активацией TGF-рецептора I типа, мы использовали ингибитор киназного домена TRI (SB-505124).
В ранее опубликованных работах была определена максимально эффективная для клеток HepG2 доза ингибитора SB-505124, при которой ингибитор специфически подавляет функцию киназного домена TRI [DaCosta Byfield et al., 2004]. Совместно с Д.В. Альперном были проведены эксперименты в которых, клетки гепатомы HepG2 обрабатывали ингибитором киназного домена SB-505124 в концентрации 1 мкМ вместе с цитокином TGF2 (5 нг/мл). Клетки инкубировали 24 часа, фиксировали и проводили иммунофлуоресцентное окрашивание специфическими антителами к различным группам изоформ белка HNF4. Мы обнаружили, что ингибирование киназного домена TRI блокирует TGF-зависимую активацию экспрессии HNF4P2 и подавление экспрессии HNF4P1 (Рис. 3).
Таким образом, можно заключить, что действие, оказываемое TGF2 на экспрессию двух групп изоформ HNF4, осуществляется посредством передачи сигнала именно через рецептор TRI, который способен активировать внутриклеточные каскады белковых взаимодействий, приводящих к изменению уровня транскрипции генов-мишеней.

Рисунок 3. Ингибитор киназного домена TRI блокирует TGF2-зависимое переключение экспрессии изоформ HNF4. Иммунофлуоресцентное окрашивание клеток HepG2 при обработке TGF2 (5 нг/мл) и/или ингибитором TRI. Справа - окраска с помощью антител к HNF4Р2, слева - к HNF4Р1.
1.3. Активация Smad сигнального каскада в клеточной линии HepG2 под действием TGF2
TGF на поверхности клетки связывается с рецептором II типа, такой комплекс связывает рецептор I типа и фосфорилирует его. Дальнейшая передача сигнала происходит при активации различных внутриклеточных субстратов фосфорилированным рецептором I типа. Классическим сигнальным каскадом, активируемым в ответ на действие цитокинов TGF, является Smad каскад.
Для того чтобы проверить, активируется ли Smad сигнальный каскад в клетках HepG2 при действии TGF, мы провели трансфекцию генетической конструкцией, в которой репортерный ген люциферазы находится под контролем нескольких Smad-связывающих элементов (CAGA). Через сутки после трансфекции клетки инкубировали с цитокинами TGF1 или TGF2 еще в течение 24 ч, а затем лизировали и измеряли активность репортерных генов люциферазы в полученных образцах. Полученные результаты указывают на почти 4-х кратное увеличение относительной люциферазной активности при действии как TGF1, так и TGF2 (Рис. 4). Таким образом, Smad–зависимый сигнальный каскад функционально активен в клетках культуры HepG2. Возможно, некоторые из наблюдаемых нами эффектов TGF1 и TGF2 реализуются посредством активации Smad белков.

Рисунок 4. Воздействие TGF1 и TGF2 приводит к индукции активности Smad каскада в клетках HepG2. Активность репортерного гена люциферазы в клетках HepG2 под контролем Smad-связывающих элементов (SBE). HepG2 – контрольные клетки, не подвергшиеся обработке, HepG2+TGF2 – клетки HepG2 после обработки TGF2. Обработку цитокином в количестве 5 нг/мл проводили в течение 24 ч через сутки после трансфекции.
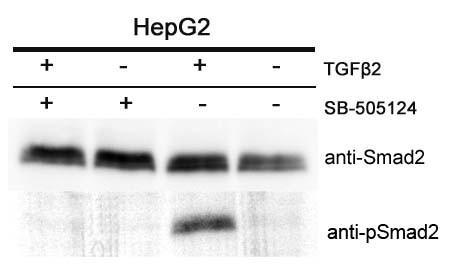
Рисунок 5. Подавление фосфорилирования Smad2 под действием ингибитора TRI. Иммуноблоттинг лизатов клеток, обработанных TGF2 и ингибитором TRI. Дорожка 1 - HepG2 после одновременной обработки TGF2(5 нг/мл) и ингибитором TRI (1 мкМ) в течение 24 ч, дорожка 2 - HepG2 после обработки ингибитором TRI в течение 24 ч, дорожка 3 – HepG2 после обработки TGF2 в течение 24 ч, дорожка 4 – контрольная линия HepG2. Верхний ряд - иммуноблоттинг с антителами к общему Smad2. Нижний ряд – иммуноблоттинг с антителами к фосфорилированной форме Smad2.
Для подтверждения полученных результатов, а также для того, чтобы проверить, произойдет ли подавление фосфорилирования Smad белков при инкубации с селективным ингибитором TRI, мы провели сравнение уровней фосфорилированного Smad2 и общего Smad2 в клеточных лизатах HepG2, а также HepG2, обработанной TGF2, и HepG2, инкубированной со специфическим ингибитором киназного домена TRI (SB-505124), методом Вестерн-блот анализа (Рис. 5). Иммуноблоттинг с антителами к фосфорилированной форме Smad2, показал, что при обработке HepG2 цитокином TGF2 фосфорилирование Smad2 значительно повышается, при этом использование ингибитора SB-505124 полностью предотвращает такое фосфорилирование.
1.4. Изменение экспрессии изоформ HNF4P1 при ингибировании МЕК сигнального пути
Как уже было отмечено выше, цитокины семейства TGF могут активировать не только канонический каскад Smad белков, но и другие сигнального пути. В экспериментах на различных клеточных линиях описана TGF –зависимая активация Erk1/2, JNK и p38, PI3K, а также Ras и Rho-подобных малых ГТФаз.
Ранее было описано участие компонентов МАР-киназного сигнального пути в регуляции экспрессии HNF4 [Hatzis et al., 2006]. Авторы сообщают, что в клетках гепатомы человека HepG2 активация МАР-киназного каскада при воздействии форболового эфира (PMA) приводит к снижению экспрессии HNF4P1.
Учитывая описанные факты, мы решили проверить, активируется ли под действием TGF1 MEK/Erk сигнальный каскад, и участвуют ли компоненты этого сигнального пути в TGF-зависимой регуляции транскрипции различных групп изоформ HNF4 в исследуемой системе.
Совместно с Д.В. Альперном клетки линии HepG2 обрабатывали специфическим ингибитором MEK PD 98059 в концентрации 20 мкМ, а также цитокином TGF2 (5 нг/мл). Ингибитор PD 98059 блокирует МАР киназы и, соответственно, передачу сигнала от рецептора TGF к киназе Erk, которая фосфорилирует транскрипционные факторы и регулирует транскрипцию генов-мишеней. В качестве положительного контроля фосфорилирования Erk2 киназы использовали клетки HepG2, обработанные форболовым эфиром. Полученные клеточные лизаты анализировали с помощью Вестерн блоттинга со специфическими антителами к фосфорилированным формам Erk2/Erk1 (Рис. 6). Таким образом мы показали, что обработка TGF2 приводит к фосфорилированию Erk2 и этот сигнальный каскад в клеточной линии HepG2 активен.
Для того чтобы проверить, какие из индуцируемых TGF2 изменений экспрессии групп изоформ HNF4 опосредованы активацией MEK/Erk сигнального каскада, проводили окрашивание клеток HepG2 специфическими антителами к двум группам изоформ HNF4P2 и HNF4P1. Мы обнаружили, что ингибирование киназы Erk приводит к подавлению лишь одного из двух эффектов, оказываемых цитокином TGF2 на активность промоторов HNF4, а именно препятствует ингибированию активности промотора Р1. TGF-зависимая индукция синтеза «эмбриональных» изоформ HNF4P2 в клетках гепатомы человека HepG2 при ингибировании МАР киназы MEK сохраняется (Рис. 7).

Рисунок 6. TGF2 опосредованная активация MEK/Erk сигнального каскада. Иммуноблоттинг лизатов клеток, обработанных TGF2, ингибитором МЕК и РМА. Дорожка 1 – контрольная линия HepG2, дорожка 2 - HepG2 после обработки TGF2 (5 нг/мл), дорожка 3 - HepG2 после обработки ингибитором МЕК (20 мкМ), дорожка 4 - HepG2 после одновременной обработки TGF2 и ингибитором МЕК, дорожка 5 – HepG2 после обработки РМА, дорожка 6 – HepG2 после одновременной обработки РМА и ингибитором МЕК. Верхний ряд - иммуноблоттинг с антителами к фосфорилированным формам Erk1/2, средний ряд – с антителами к общему Erk1/2, нижний ряд –иммуноблоттинг с антителами к -тубулину.

Рисунок 7. Ингибирование киназы МЕК приводит к блокированию TGF2-зависимого подавления синтеза HNF4Р1, но не влияет на активацию HNF4Р2. Иммунофлуоресцентное окрашивание клеток HepG2 при обработке TGF2 (5 нг/мл) и/или ингибитором MEK (PD 98059). Справа окраска антителами к HNF4Р2, слева - к HNF4Р1.
Таким образом, действие, которое оказывает индукция TGF сигнального пути на экспрессию различных групп изоформ гена HNF4, опосредовано запуском нескольких сигнальных путей. Активность промотора Р1 в исследуемой системе регулируется при участии МАР-киназ, в то время как за регуляцию промотора Р2 отвечают, по всей видимости, другие сигнальные каскады.
1.5. Анализ экспрессии генов в клеточной линии HepG2, гиперэкспрессирующей TGF2
Известно, что цитокины семейства TGF синтезируются в виде гомодимерных предшественников. В результате процессинга во внеклеточную среду секретируется активная форма TGF, связанная с отщепившимся полипептидом, который называется LAP. Считается, что в таком виде TGF не связывается со своим рецептором и поэтому неактивен. Однако при воздействии ряда факторов (протеазы, интегрины, изменения рН, активные формы кислорода) TGF активируется и взаимодействует с рецепторными системами.
Ранее в нашей лаборатории при определении количества TGF2 в культуральной среде клеточной линии Н33, полученной из быстрорастущей гепатокарциномы, с помощью метода ИФА (иммуноферментный анализ) было обнаружено, что большая часть TGF2 находится в латентной (непроцессированной) форме.
Для исследования вклада активной и латентной форм TGF2 в регуляцию процессов, ассоциированных с прогрессией ГК, а также для изучения эффектов долговременного действия TGF2 нами были получены клеточные линии HepG2, гиперэкспрессирующие нормальную или мутантную формы TGF2. Одна из использованных в этой работе экспрессиурующих конструкций кодировала нормальный ген TGF2 человека (pLVTGF2N), другая - мутантный вариант TGF2 (pLVTGF2RG299). Эта мутация, в результате которой аргинин в положении 299 заменен на глицин, приводит к нарушению внутриклеточного процессинга, и во внеклеточную среду секретируется конститутивно латентный белок.
Клеточная линия HepG2 была инфицирована лентивирусами, кодирующими описанные варианты TGF2. После селекции на блеомицине были отобраны клоны, гиперэкспрессирующие нормальный или конститутивно латентный TGF2. Для последующих экспериментов были выбраны клоны, экспрессирующие максимальное количество цитокина: клон HepG2 TGF2 N cl1 и клон HepG2 TGF2 R299G cl5, секретирующий мутантную форму TGF2. В качестве контрольной линии использовали неинфицированные клетки HepG2, продуцирующие TGF2 в количестве порядка 1 нг/млн клеток (Таб. 1).
Методом ОТ-ПЦР мы исследовали экспрессию некоторых гепатоспецифических генов и генов, ассоциированных с прогрессией ГК, в клетках HepG2, гиперэкспрессирующих TGF2, и в контрольной культуре.
| TGF2 нг/106 клеток | |
| HepG2 | 1.1 |
| HepG2 TGF2 N cl1 | 13 |
| HepG2 TGF2 R299G cl5 | 67 |
Таблица 1. Количество TGF2 в кондиционированной среде культур HepG2, гиперэкспрессирующих нормальную или мутантную формы TGF2. Результаты определения общего количества белка TGF2 методом ИФА в кондиционированной среде HepG2, HepG2 TGF2 N cl1 и HepG2 TGF2 R299G cl5. Результаты определения общего количества TGF2 представлены в нг на 1х106 клеток.

Мы обнаружили, что при гиперэкспрессии как мутантной, так и нормальной форм TGF2 происходит активация экспрессии транскрипционного фактора Snail и репрессия гена Е-кадхерина, одного из ключевых кадхеринов, обеспечивающих гомотипическую адгезию в эпителиальных тканях (Рис. 8). Snail способен связываться с промоторной областью гена Е-кадхерина, подавляя экспрессию последнего [Cano et al., 2000]. Таким образом, можно предположить, что в исследуемой модели происходит Snail-зависимое подавление экспрессии Е-кадхерина. Snail-зависимая репрессия транскрипции гена Е-кадхерина индуцирует ЭМП в опухолях эпителиального происхождения, способствуя таким образом приобретению опухолевыми клетками инвазивного фенотипа и способности к метастазированию [Cicchini et al., 2006].
Рисунок 8. Гиперэкспрессия TGF2 в клеточной линии HepG2 приводит к подавлению экспрессии некоторых гепатоспецифических генов и генов, ассоциированных с прогрессией ГК. ОТ-ПЦР анализ экспрессии генов в клеточной линии HepG2, гиперэкспрессирующей нормальную или мутантную формы TGF2. Дорожка 1 – HepG2 TGF2 N cl1, дорожка 2 – HepG2 TGF2 R299G cl5, дорожка 3 – контроль - HepG2. В качестве контроля равенства количества РНК, взятой в реакцию, приведены результаты ОТ-ПЦР с праймерами к GAPDH.
Кроме того, при гиперэкспрессии TGF2 в клетках HepG2 было выявлено подавление экспрессии бетагликана (Рис. 8). Бетагликан или рецептор TGF III типа (TRIII) не имеет внутриклеточного киназного домена и, соответственно, не способен участвовать во внутриклеточной передаче сигнала, но принимает участие в активации и накоплении TGF на поверхности клеток и модулирует его связывание с сигнальными рецепторами TRI и TRII. Сниженная по сравнению с нормальными тканями экспрессия TRIII или отсутствие таковой описаны в опухолях молочной железы, яичников, легких и поджелудочной железы, более того, это снижение коррелирует с гиперэкспрессией TGF и со стадией прогрессии опухоли. Ряд авторов предполагает возможную противоопухолевую роль этого белка, так как он способен блокировать действие цитокинов семейства TGF [Gordon et al., 2007; Hempel et al., 2007; Finger et al., 2008]. Необходимо отметить, что экспрессия генов остепонтина, фибронектина и 3 субъединицы интегрина, транскрипция которых повышалась при обработке TGF2 культуры HepG2, при гиперэкспрессия этого цитокина в культуре HepG2 не изменилась.
Повышение продукции TGF2 вызвало снижение уровня экспрессии некоторых гепатоспецифических генов (Рис. 8). Мы обнаружили снижение суммарного количества HNF4, что явилось следствием подавления активности обоих промоторов гена HNF4. Кроме того, подавление экспрессии обеих групп изоформ вызвало снижение экспрессии непосредственной транскрипционной мишени HNF4P1 – гена АроАIV (Рис. 8).
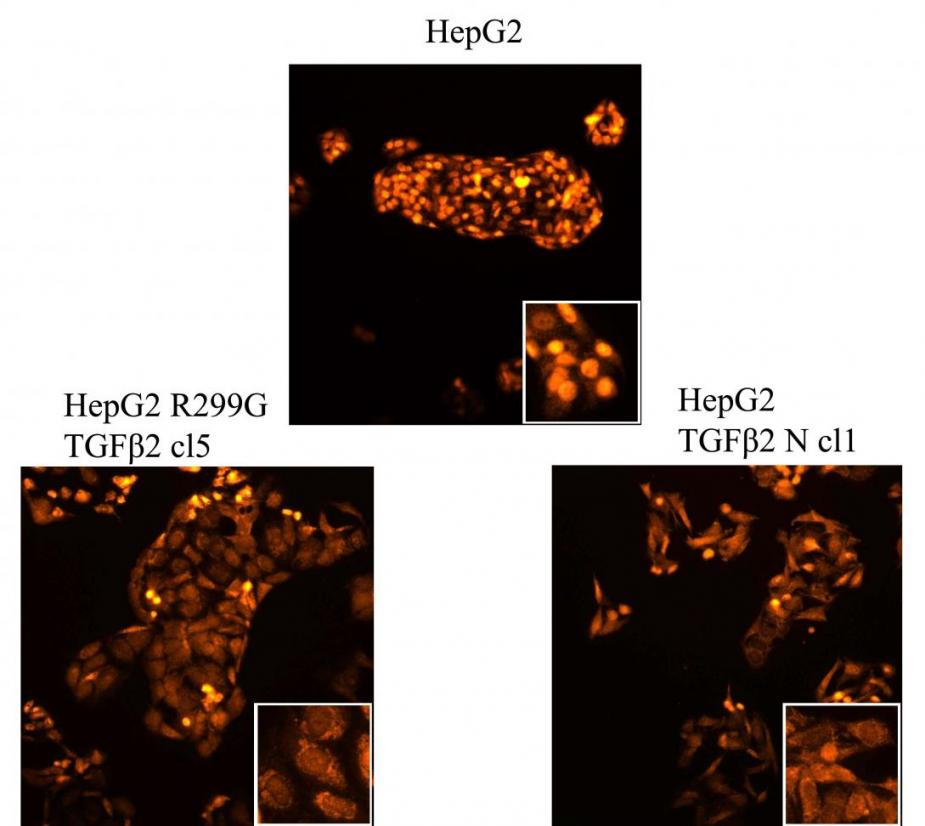
Рисунок 9. Подавление продукции HNF4 при гиперэкспрессии TGF2 в клеточной линии HepG2. Иммунофлуоресцентное окрашивание клеток HepG2, гиперэкспрессирующих нормальную и мутантную формы TGF2. Окрашивание антителами к HNF4. Сверху (контроль) - HepG2, слева - HepG2 TGF2 R299G cl5, справа - HepG2 TGF2 N cl1.
Подавление синтеза HNF4 в клетках HepG2, гиперэкспрессирующих нормальную и мутантную формы TGF2, наблюдалось также при иммунофлуоресцентном окрашивании клеток с помощью антител к общим доменам различных групп изоформ HNF4. В контрольном препарате HepG2 примерно 77 % клеток демонстрирует ядерную окраску на HNF4, однако при гиперэкспрессии TGF2 происходит снижение количества клеток с окрашенными ядрами. В клетках, экспрессирующих нормальную форму TGF2, окрашенные ядра составляют 20 %, а в клетках, которые экспрессируют мутантную форму TGF2 - 25 % (Рис. 9).
1.6. Изменение пролиферативных характеристик клеточной линии HepG2 при гиперэкспрессии TGF2
Для сравнения кинетики роста клеточной линии HepG2 и ее производных мы использовали метод прямого подсчета клеток. Оказалось, что на 6 день культивирования количество клеток в культурах, гиперэкспрессирующих как нормальную, так и мутантную формы TGF2, увеличилось в два раза по сравнению с контрольной культурой HepG2 (Рис. 10А).
Для определения скорости пролиферации использовали метод измерения включения модифицированного нуклеотида – 5-бромдезоксиуридина (5-BrdU) в ДНК с последующей иммунофлуоресцентной окраской специфическими антителами к модифицированному нуклеотиду. После окраски проводили подсчет общего количества клеток и клеток, включивших метку, в поле зрения. Результаты этого эксперимента представлены как отношение клеток, включивших метку, к общему количеству клеток. При гиперэкспрессии мутантной и нормальной форм TGF2 количество клеток, включивших 5-BrdU, примерно на 15 % и 10 %, соответственно, превышало этот показатель в контрольной культуре HepG2 (Рис. 10Б).
Для изучения способности клеток выживать и формировать колонии в условиях большого разведения был проведен тест на клоногенную активность. Клетки исследуемых линий рассевали при низкой плотности (около 1500 клеток на 3 см чашку) и культивировали в течение 2 недель. После фиксации клетки окрашивали гематоксилином и сравнивали количество выросших макроколоний в экспериментальных и контрольных чашках. Из приведенных фотоизображений видно, что клетки контрольной культуры HepG2 способны дать начало меньшему количеству клонов, чем культуры, гиперэкспрессирующие нормальную или мутантную формы TGF2 (Рис. 11). Таким образом, при повышении продукции TGF2 в гепатоме HepG2 способность клеток выжить и размножиться в условиях большого разведения по сравнению с контрольной культурой HepG2 увеличивается.
Обобщая данные, полученные в двух сериях экспериментов, можно сделать вывод о том, что TGF2 вызывает различные эффекты в зависимости от того, в какой форме он связывается с мембранными рецепторами на поверхности клеток HepG2. При инкубации с активным рекомбинантным димером TGF2 транскрипция изоформ гена HNF4, преобладающих во взрослой печени, снижается, а уровни «эмбриональных» изоформ, наоборот, повышаются. Однако при экзогенной экспрессии гена TGF2 в этой же клеточной системе мы наблюдали иную картину: экспрессия гена HNF4 подавляется, причем снижение затрагивает обе группы изоформ. Кроме того, различия выражались не только в экспрессии изоформ гена HNF4, но и в спектре экспрессии различных TGF-индуцируемых генов. Так, например, если в первой экспериментальной системе транскрипция гена Snail не изменилась, но повысилась экспрессия остеопонтина, фибронектина и 3 интегрина, то во второй модели, наоборот, экспрессия Snail увеличилась, а транскрипция остеопонтина и фибронектина осталась неизменной. Возможно, что эти различия обуславливаются тем, что рекомбинантный TGF2 представляет собой активный димер, а в клетках HepG2 при гиперэкспрессии цитокина процессинга не происходит. Так как при определении количества TGF2 с помощью метода ИФА большая часть TGF2 определяется только после снижения рН образцов (как HepG2 гиперэкспрессирующей нормальный вариант TGF2, так и мутантный), что указывает на отсутствие в этих образцах активного димера.
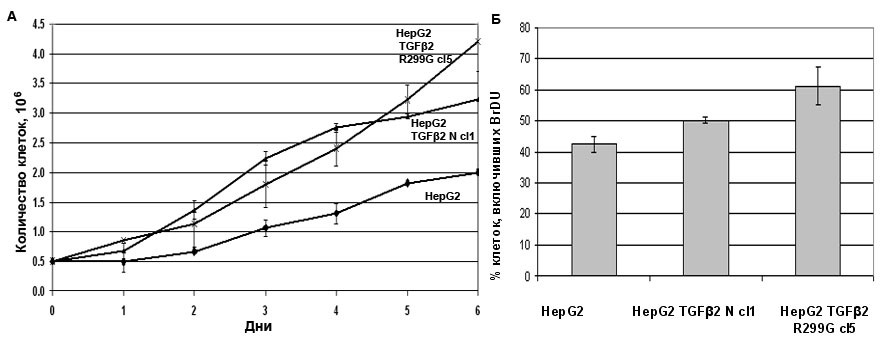
Рисунок 10. Гиперэкспрессия TGF2 повышает скорость пролиферации HepG2. А. Сравнение кинетики пролиферации клеток HepG2, гиперэкспрессирующих нормальную и мутантную формы TGF2. Б. Скорость пролиферации клеток HepG2, гиперэкспрессирующих нормальную или мутантную формы TGF2, оцененная по включению 5-BrdU.

Рисунок 11. При гиперэкспрессии нормальной или мутантной формы TGF2 в культуре HepG2 способность образовывать клоны увеличивается. Результаты окраски гематоксилином клонов, выросших через 2 недели культивирования из 1500 клеток линии HepG2, HepG2 TGF2 N cl1, HepG2 TGF2 R299G.
2. Изучение эффектов, вызванных подавлением продукции TGF2 в клетках дедифференцированной гепатомы Н33
2.1. Подавление продукции TGF2 в клеточной линии Н33
Для подтверждения возможной роли TGF2 в качестве фактора, способствующего прогрессии ГК, необходимо было наряду с моделями, на которых изучались эффекты, индуцируемые TGF2, рассмотреть модель, в которой можно было бы наблюдать эффекты, обусловленные подавлением активности TGF2.
В качестве такой модельной системы была выбрана клеточная линия Н33, полученная ранее из быстрорастущего дедифференцированного варианта ГК мыши [Кудрявцева и соавт., 2000]. В культуре Н33 повышена экспрессия многих TGF-индуцируемых генов, экспрессия которых характерна для злокачественных опухолей (EDA+ сплайс формы фибронектина 1, остеопонтина, 3 субъединицы интегрина, Snail, виментина), а экспрессия генов, характерных для дифференцированных гепатоцитов, наоборот, подавлена. Необходимо отметить также, что в ходе одноступенчатой прогрессии повысилась экспрессия гена TGF2, в то время как экспрессия гена TGF1 не изменилась.

Рисунок 12. Снижение количества TGF2 в кондициони-рованной среде Н33-миTGF2-1 и Н33-миTGF2-2. Результаты определения общего количества TGF2 методом ИФА представлены в виде процентного соотношения между количеством TGF2, секретируемого контрольной культурой, и количеством TGF2 в кондиционированной среде линий Н33-миTGF2-1 и Н33-миTGF2-2.
Для того чтобы подавить синтез TGF2 в культуре Н33, мы использовали лентивирусы, кодирующие два типа малых интерферирующих РНК к гену TGF2 (миTGF2-1 и миTGF2-2). В качестве контрольной культуры были использованы клетки Н33, инфицированные тем же вектором, не содержащим миРНК (Н33-pLSL-puro). После инфекции проводилась селекция трансдуцированных клеток на пуромицине.
Уровни секреции белка TGF2 в клетках Н33, трансформированных pLSL-puro-миTGF2-1 (Н33-миTGF2-1), pLSL-puro-миTGF2-2 (Н33-миTGF2-2), и в контрольной культуре Н33-pLSL-puro определяли методом ИФА. На Рис. 12 представлена диаграмма, которая отражает падение уровня продукции TGF2 в культурах, экспрессирующих миРНК TGF2, в процентном отношении по сравнению с контрольной культурой Н33-pLSL-puro. Оказалось, что две последовательности миРНК в разной степени подавляют экспрессию TGF2: в культуре Н33-миTGF2-1 регистрировалось примерно трехкратное снижение продукции по сравнению с контролем, в то время как в культуре Н33-миTGF2-2 произошло подавление лишь в 1.5 раза. Эти данные подтверждают результаты ОТ-ПЦР со специфическими праймерами к гену TGF2 (Рис. 13). Необходимо отметить, что последовательности миРНК TGF2 были специфичны и не повлияли на уровень транскрипции гена TGF1 (Рис. 13).
2.2. Изменения экспрессии генов в клеточной линии Н33, трансфицированной миРНК к TGF2
По данным ОТ-ПЦР, в линии Н33-миTGF2-1, где продукция TGF2 максимально снижена по сравнению с контрольными клетками и клетками Н33-миTGF2-2, наблюдается активация транскрипции гена HNF4, кроме того, повышается уровень экспрессии C/EBP и бетагликана. (Рис. 13)

Уровень транскрипции гена HNF4 в культуре дедифференцированной гепатомы Н33 при подавлении синтеза TGF2 остается достаточно низким и, вследствие этого, методом ОТ-ПЦР нельзя получить информацию о транскрипции его изоформ.
Рисунок 13. Влияние подавления продукции TGF2 на экспрессию генов, регулирующих дифференцировку гепатоцитов, в клеточной линии Н33. Результаты ОТ-ПЦР: дорожка 1 – Н33-pLSL-puro, дорожка 2, 3 – Н33-миTGF2-1 и Н33-миTGF2-2, экспрессирующие миРНК к TGF2. Для контроля равенства количества РНК, взятой в реакцию, приведены результаты ОТ-ПЦР с праймерами к GAPDH.
Невысокое, но достоверное восстановление экспрессии гена HNF4 при подавлении продукции TGF2 в клетках дедифференцированной гепатомы Н33, является важным результатом, указывающем на возможную роль активации гена TGF2 в прогрессии ГК. В предыдущих работах нашей лаборатории на различных системах, в том числе при анализе изменения уровней транскрипции генов опухолевых образцах ГК человека, было показано, что снижение экспрессии HNF4 определяет уровень дифференцировки опухолей и коррелирует со стадией прогрессии ГК [Флейшман, 2008]. Также было продемонстрировано, что в культуре Н33 ген HNF4 инактивирован не за счет мутации или делеции, а за счет транскрипционной регуляции, предположительно – репрессором, который связывается с определенным районом в промоторе Р1 этого гена.
2.3. Изменение скорости пролиферации культуры клеток Н33 при подавлении продукции TGF2
Подавление синтеза TGF2 привело к невысокой, но достоверной реэкспрессии HNF4 и C/EBP, факторов, которые не только влияют на дифференцировку, но и участвуют в контроле пролиферации гепатоцитов и некоторых других типов эпителиальных опухолей.

Учитывая эти факты, мы исследовали скорость пролиферации в клетках Н33 с помощью метода включения 5-BrdU в ДНК. В контрольной культуре Н33-pLSL-puro примерно 70 % клеток были окрашены, а при подавлении синтеза TGF2 процент пролиферирующих клеток снизился до 55% в культуре Н33-миTGF2-1 и 60% в культуре Н33-миTGF2-2 (Рис.14).
Рисунок 14. Подавление продукции TGF2 в культуре Н33 приводит к снижению скорости пролиферации клеток. Скорость пролиферации клеточных линий Н33-pLSL-puro, Н33-миTGF2-1 и Н33-миTGF2-2, определенная по включению модифицированного нуклеотида 5-BrdU в ДНК.
Таким образом, подавление продукции TGF2 в культуре H33 привело к снижению скорости пролиферации клеток. Более того, уровень такого снижения коррелировал со степенью подавления продукции TGF2, то есть в культуре, где миРНК TGF2 более эффективно подавили синтез TGF2, замедление пролиферации оказалась более значительным по сравнению с линией Н33-миTGF2-2, где экспрессировалась менее эффективная последовательность миРНК TGF2.
Вероятно, эффект подавления пролиферации может быть обусловлен как реэкспрессией HNF4 и C/EBP, так и непосредственно подавлением продукции TGF2, так как цитокины семейства TGF, как было отмечено ранее, могут индуцировать клеточный цикл посредством различных механизмов [Derynck and Zhang, 2003; Sato et al., 2008].
2.4. Исследование подвижности клеток H33 при подавлении продукции TGF2
Поскольку известно, что TGF1 участвует в регуляции инвазии и подвижности различных типов эпителиальных клеток [Hempel et al., 2007], мы решили исследовать, влияет ли подавление продукции TGF2 на подвижность клеток дедифференцированной гепатомы H33.
При постановке теста на определение клеточной подвижности мы использовали инвазионные камеры с мембранами, содержащими поры. В верхнюю часть камеры наливали среду без сыворотки с клетками, в лунку плашки помещали среду с 5% содержанием сыворотки. Таким образом, клетки по градиенту концентрации сыворотки проползали через мембрану с порами 8 мкм. Через сутки клетки, которые остались на верхней стороне мембраны, удаляли, а клетки, которые проползли сквозь поры, окрашивали и подсчитывали.
Мы обнаружили, что в культуре Н33-миTGF2-1, где подавление TGF2 было наиболее значительным, произошло резкое снижение подвижности клеток: количество клеток, которое прошло через мембрану, было в 7 раз меньше аналогичного показателя для контрольной культуры Н33-pLSL-puro. Подвижность клеток культуры Н33-миTGF2-2 снизилась в 3 раза по сравнению с контролем (Рис. 15).

Рисунок 15. Подавление продукции TGF2 в культуре Н33 вызывает снижение подвижности клеток. Результаты представлены в виде процентного отношения количества клеток Н33-миTGF2-1 и Н33-миTGF2-2 к количеству контрольных клеток Н33-pLSL-puro, которые проползли через мембрану (8 мкм) по градиенту сыворотки в среде. Результаты получены в трех независимых экспериментах.
Способность TGF1 индуцировать клеточную подвижность была продемонстрирована ранее на культурах клеток карцином яичников и поджелудочной железы [Hempel et al., 2007]. Более того, при воздействии TGF1 в этих культурах наблюдалось подавление экспрессии бетагликана, а реэкспрессия этого гена приводила к снижению клеточной подвижности. Так как в исследуемой системе наблюдается снижение подвижности клеток и повышение экспрессии бетагликана, можно предположить, что бетагликан также вносит вклад в изменение клеточной подвижности в этой системе.
ЗАКЛЮЧЕНИЕ
В представленной работе было проведено исследование возможной роли TGF2 в прогрессии ГК. Для этого были использованы две экспериментальные системы: линия дифференцированной гепатомы человека HepG2, в которой не экспрессируются цитокины семейства TGF, и полученная из низкодифференцированной быстрорастущей ГК мыши культура Н33, в которой нами выявлена гиперэкспрессия TGF2. Таким образом, обрабатывая цитокином TGF2 или индуцируя его экзогенную экспрессию в гепатоме HepG2, и, наоборот, подавляя его продукцию в дедифференцированной гепатоме Н33, мы предполагали выяснить вклад TGF2 в прогрессию ГК.
В клетках дифференцированной гепатомы HepG2 обработка как TGF1, так и TGF2 привела к повышению экспрессии генов 3 субъединицы интегрина, остеопонтина, фибронектина и ig-h3, для которых показана активация в ходе прогрессии ГК. Кроме того, произошло снижение экспрессии генов, кодирующих ГЯФ, которые определяют дифференцировку гепатоцитов. Одним из таких факторов является HNF4 – центральное звено гепатоспецифической регуляторной сети. При обработке TGF1 и TGF2 произошло координированное изменение экспрессии двух групп изоформ этого гена: снижение экспрессии группы, характерной для дифференцированных гепатоцитов, и повышение экспрессии «эмбриональных» изоформ. Мы также показали, что внутриклеточные механизмы этих изменений различны: снижение HNF4Р1 происходит за счет TGF-зависимой активации MEK – Erk сигнального пути, при этом повышение HNF4Р2 опосредовано другим механизмом.
Ранее в нашей лаборатории было показано, что большая часть TGF, который продуцируют клетки дедифференцированной гепатомы мыши Н33, находится в латентной (непроцессированной) форме. Для исследования вклада активной и латентной форм TGF2 в регуляцию процессов, ассоциированных с прогрессией ГК, а также для изучения эффектов долговременного действия TGF2 нами были получены клеточные линии HepG2, гиперэкспрессирующие нормальную или мутантную формы TGF2. Мутация в гене TGF2 приводила к нарушению процессинга, и в среду секретировался конститутивно латентный белок.
В клетках HepG2, гиперэкспрессирующих нормальный и мутантный вариант TGF2, произошло снижение экспрессии гена Е-кадхерина и активация транскрипции гена, кодирующего его репрессор – Snail. Кроме того, произошло снижение экспрессии двух групп изоформ гена HNF4 и непосредственной мишени HNF4Р1 – гена ApoAIV. Необходимо отметить, что гиперэкспрессия TGF2 привела к ускорению пролиферации и к повышению клоногенной активности этих клеток.
Подавление продукции TGF2 в клетках Н33 привело к реактивации транскрипции генов, экспрессия которых критична для поддержания дифференцировки гепатоцитов и нарушение функции которых описано для ГК - HNF4 и С/ЕВР. Более того, снижение синтеза TGF2 привело к снижению таких ключевых для опухолевых клеток характеристик, как скорость пролиферации и подвижность. Все это свидетельствует в пользу того, что при подавлении продукции TGF2 в культуре Н33 произошла частичная реверсия злокачественного фенотипа опухолевых клеток.
Итак, мы показали, что TGF2 снижает экспрессию генов, продукты которых контролируют дифференцировку гепатоцитов, и усиливает пролиферацию и подвижность клеток, способствуя тем самым развитию более злокачественного фенотипа ГК. Таким образом, разработка методов эффективного подавления продукции этого цитокина как in vitro, так и in vivo представляется перспективным направлением в развитии методов противоопухолевой терапии.
ВЫВОДЫ
- В клетках гепатомы человека HepG2 воздействие цитокинов TGF1 и TGF2 вызывает подавление экспрессии факторов, контролирующих дифференцировку гепатоцитов (C/EBP, HNF1), и активацию генов, продукты которых ассоциированы с прогрессией ГК (остеопонтин, фибронектин и 3 субъединица интегрина).
- При обработке TGF1 и TGF2 в клетках гепатомы человека HepG2 происходит координированное изменение активности двух альтернативных промоторов гена HNF4: активация альтернативного Р2 промотора и подавление основного промотора Р1. TGF-зависимое подавление промотора Р1 гена HNF4 опосредованно активацией МАР-киназного сигнального пути.
- Экспрессия экзогенного TGF2 в клетках HepG2 снижает экспрессию ключевых регуляторов гепатоцитарной дифференцировки C/EBP и HNF4.
- При гиперэкспрессии TGF2 происходит ускорение пролиферации и повышение клоногенной активности клеток гепатомы HepG2.
- При подавлении продукции TGF2 в клетках дедифференцированной гепатомы Н33 происходит реактивация эндогенной экспрессии факторов HNF4 и С/ЕВР.
- Подавление продукции TGF2 в клетках Н33 вызывает снижение подвижности и скорости пролиферации этих клеток.
- TGF2 способен влиять на дифференцировочные, пролиферативные и миграционные свойства опухолевых клеток при прогрессии гепатоцеллюлярных карцином.
СПИСОК РАБОТ, ОПУБЛИКОВАННЫХ ПО ТЕМЕ ДИССЕРТАЦИИ
- Д.В. Альперн, М.В. Макарова, О.А. Чернобельская, Д.И. Флейшман, Н.Л. Лазаревич. р53 и TGF регулируют экспрессию эмбриональной изоформы Гепатоцитарного Ядерного Фактора 4 в клетках гепатомы человека. (2008) Российский Биотерапевтический Журнал, том 7(3): 56–69.
- М.В. Макарова, Н.Е. Доннер, Д.И. Флейшман, О.В. Морозова, Н.Л. Лазаревич. Частичная реверсия злокачественного фенотипа клеток дедифференцированной гепатокарциномы мыши инактивацией трансформирующего фактора роста 2. (2009) Российский Биотерапевтический Журнал, том 3(3): 56-59.
- M.V. Makarova, N.E. Donner, D.I. Fleishman, O.V. Morozova, N.L. Lazarevich. Role of TGF2 in Hepatocellular Carcinoma Progression. 44th Annual Meeting of the European Association for the Study of the Liver, EASL, Copenhagen, Journal of Hepatology, 2009,, Vol. 50, Sup. 1, S196.
- M.V. Makarova, D.V. Alpern, N.E. Donner, N.L. Lazarevich. Differential regulation of nuclear receptor HNF4 isoforms expression by TGF in HepG2 Human hepatoma cells. EASL – AASLD Conference «Nuclear receptors and Liver desease», Vienna, February 27-March 1, 2009. Abstract book. P. 80.
- N.L. Lazarevich, D.I. Fleishman, O.A. Chernobel’skaya, M. V. Makarova, D.V. Alpern, O.V. Morozova, Yu.I. Patyutko, N.V. Engelhardt. Tissue-specific transcription factor network in hepatocellular carcinoma progression. The European Cancer Conference ECCO 13, Paris, Eur. J. Cancer, 2005, v.3, No 2 (Suppl.), p. 215.
- Макарова М.В., Флейшман Д.И., Морозова О.Д., Овчинников Д.А., Лазаревич Н.Л. Активация TGF сигнального пути при прогрессии гепатокарцином. Материалы VI Международной конференции «Молекулярная генетика соматических клеток». 12-16 декабря 2005 г., Звенигород, Москва, стр. 45.
- M.V. Makarova, O.A. Chernobelskaya, D.I. Fleishman, O.V. Morozova, N.L. Lazarevich. Role of TGF-b signaling in hepatocellular carcinoma progression. FEBS Special Meeting on Cellular Signaling, May 26 - June 1, 2006, Dubrovnik, Croatia. Abstract book, p.146-147.
- D.I. Fleishman, M.V. Makarova, D.V. Alpern, D.A. Shavochkina, A.A. Kuznetzova, O.V. Morozova, Y.I. Patyutko, N.L. Lazarevich. Gene expression alterations in hepatocarcinogenesis. Molecular Mechanisms in Signal Transduction and Cancer. FEBS/EMBO Special meeting, August 15-24, 2007, Spetses, Greece. Abstract book, p 28.
- D. Alpern, M. Makarova, D. Fleishman, O. Chernobelskaya, N. Lazarevich Expression of HNF4alpha is switched under influence of TGFbeta and p53. United Kingdom, Bath, 26-28 June, 2007. JDRF Center for Beta Cell Therapy in Diabetes Training Course "Transdifferentiation to Beta Cells", pp 16-17.
- N. Lazarevich, D. Fleishman, O. Chernobelskaya, M. Makarova, D. Alpern, A. Kuznetsova, D. Shavochkina, O. Morozova, Y. Patyutko. Dysfunction of tissue-specific transcriptional network is an important determinant of hepatocellular carcinoma progression Budapest, Hungary 1-4 July 2006. EACR-19 Meeting Proceedings, 174.
- D.A. Shavochkina, D.I. Fleyshman, M.V. Makarova, A.G. Kuznetzova, O.V. Morozova, N.L. Lazarevich. Tissue-specific alterations of gene expression on the initial studies of liver carcinogenesis in mice. Spetses Summer School on Nuclear Receptor Signaling, Island of Spetses, Greece, August 26 - 31, 2007 Abstract book.