

Разработка унифицированных путей синтеза моно- и полициклических биологически активных производных 2- и 4-амино(гидразино)пиримидинов
На правах рукописи

ЕРКИН АНДРЕЙ ВИКТОРОВИЧ
РАЗРАБОТКА УНИФИЦИРОВАННЫХ ПУТЕЙ СИНТЕЗА
МОНО- И ПОЛИЦИКЛИЧЕСКИХ БИОЛОГИЧЕСКИ АКТИВНЫХ ПРОИЗВОДНЫХ 2- И 4-АМИНО(ГИДРАЗИНО)ПИРИМИДИНОВ
Специальность 02.00.03 – Органическая химия
АВТОРЕФЕРАТ
диссертации на соискание ученой степени
доктора химических наук
Санкт-Петербург
2011
Работа выполнена в федеральном государственном бюджетном образовательном учреждении высшего профессионального образования «Санкт-Петербургский государственный технологический институт (технический университет)»
ОФИЦИАЛЬНЫЕ ОППОНЕНТЫ:
Доктор химических наук, профессор Москвин Андрей Вадимович
Доктор химических наук, профессор Попова Лариса Михайловна
Доктор химических наук, профессор Илюшин Михаил Алексеевич
ВЕДУЩАЯ ОРГАНИЗАЦИЯ: Федеральное государственное унитарное предприятие «Российский научный центр «Прикладная химия»
Защита состоится « » ___________ 2012 г. в _____ час. в ауд. __ на заседании Совета по защите докторских и кандидатских диссертаций Д 212.230.02 при федеральном государственном бюджетном образовательном учреждении высшего профессионального образования «Санкт-Петербургский государственный технологический институт (технический университет)».
С диссертацией можно ознакомиться в фундаментальной библиотеке федерального государственного бюджетного образовательного учреждения высшего профессионального образования «Санкт-Петербургский государственный технологический институт (технический университет)».
Отзывы на автореферат в одном экземпляре, заверенные печатью, просим направлять по адресу: 190013, Санкт-Петербург, Московский пр., 26, ФГБОУ ВПО «Санкт-Петербургский государственный технологический институт (технический университет)». Тел.: 494-93-75, факс: 712-77-91, e-mail: [email protected]
Автореферат разослан « » ___________ 201_ г.

Ученый секретарь диссертационного совета,
кандидат химических наук, доцент Соколова Н.Б.
ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ
Актуальность темы. Производные пиримидина и, в первую очередь, компоненты нуклеиновых кислот привлекают пристальное внимание в качестве базовых объектов для создания новых биологически активных препаратов. Основой использования именно этих гетероциклических соединений в медицинской химии служат две фундаментальные гипотезы о возможности их действия как ингибиторов синтеза нуклеиновых кислот в инфицированных клетках или транспортных молекул, осуществляющих доставку фармакофорных групп к ингибируемой биологической мишени. Первое обеспечивается встраиванием фосфорилированного аналога природного пиримидина в нуклеотидную последовательность, а второе – способностью проникать через клеточные мембраны в минимально измененном виде. Примерами лекарственных препаратов, созданных с привлечением указанных гипотез, служат 5-фторурацил и 5-ди(2-хлорэтил)амино-6-метилурацил, которые применяются в химиотерапии злокачественных новообразований. Несмотря на то, что 2-амино-4(3Н)-пиримидинон – изостерический аналог цитозина – не входит, подобно урацилу, в число обязательных компонентов нуклеиновых кислот, его структура также неоднократно подвергалась химической модификации. Итогом этого явилось обнаружение двух групп биологически активных соединений: 5,6-дизамещенных изоцитозинов, сильных низкомолекулярных индукторов интерферона и специфических антивирусных агентов, и 2-гидразинопиримидинов и их производных, сравнимых по уровню противотуберкулезной активности с гидразидом (пиридин-4-ил)карбоновой кислоты («Изониазидом»). С учетом возрастающей резистентности вирусов и микобактерий туберкулеза к существующим препаратам соответствующего действия полученные результаты приобретают особое значение и могут служить отправной точкой углубленных исследований, направленных на изыскание новых биологически активных веществ среди указанных соединений. Необъяснимое отсутствие сведений о проведении таковых в литературе последних лет, с одной стороны, вызывает крайнее удивление, а с другой создает несомненный прецедент для их реализации.
Цель работы состояла в разработке унифицированных путей синтеза новых моно- и полициклических производных 2- и 4-амино(гидразино)пиримидинов и последующем изучении их биологической активности. Для достижения поставленной цели предстояло обеспечить решение следующих задач:
синтезировать реакционноспособные соединения из означенных классов гетероциклов;
исследовать трансформации синтезированных соединений под действием выбранных реагентов и достоверно установить структуру образующихся продуктов и предпочтительных форм их существования с помощью совокупного применения современных физико-химических методов анализа;
подвергнуть расширенному биологическому скринингу целевые гетероциклы с привлечением объектов бактериального, микобактериального и грибкового происхождений и выявить факторы, ответственные за проявление ими определенных видов активности.
Научная новизна.
1. Исследованы прототропная таутомерия и химические превращения 2-амино-6-метил-4(3Н)-пиримидинонов и их производных, отличные от описанных типов гетероциклизации в имидазо[1,2-a]пиримидины:
• установлен факт равновесия между 4-оксо-3,4-дигидро- и 4-гидроксиформами в водном растворе, смещающегося в сторону первой при уменьшении полярности среды;
• обнаружена внутримолекулярная перегруппировка 6-метил-2-[2-(фенилкарбамоилокси)-этил]амино-4(3Н)-пиримидинона в 2-(2-гидроксиэтил)амино-6-метил-5-фенилкарбамоил-4(3Н)-пиримидинон под действием хлороводородной кислоты;
• выявлены причины селективного N3-метилирования 2-(2-гидроксиэтил)амино-6-метил-4(3Н)-пиримидинона в растворителях-донорах водородной связи и образования смеси продуктов N3- и O-метилирования в координирующих растворителях;
• подтверждено участие атома N1 в протонировании продуктов аминирования 2-(2-ацетоксиэтил)амино-6-метил-4-хлорпиримидина ароматическими аминами в соотношении субстрат-реагент 1:1.
2. Изучены химические трансформации гидразонов 6-метилурацила, его 2-метилтиопроизводного и их аналогов, не включающие известную гетероциклизацию в 1,2,4-триазоло[4,3-a]- или -[1,5-a]пиримидины:
• осуществлен синтез 4-арилиден-1-(4-оксо-3,4-дигидропиримидин-2-ил)-5(4Н)-пиразол-онов непосредственной циклоконденсацией (4-оксо-3,4-дигидропиримидин-2-ил)гидр-азонов этилацетоацетата с ароматическими альдегидами в присутствии основания;
• установлен механизм термической деструкции (3,6-диметил-4-оксо-3,4-дигидро-пиримидин-2-ил)гидразона этилацетоацетата с трансформацией субстрата в 4,7-диметил-1,2,4-триазоло[1,5-a]пиримидин-5(4Н)-он;
• обнаружена внутримолекулярная перегруппировка 2-бензилиденгидразино-4,6-диметил-пиримидина в 2-гидразино-4-метил-6-(2-фенилэтенил)пиримидин в условиях кислотного гидролиза субстрата;
• определена последовательность ионизации 6-метил-2-(3-метил-5-оксо-2,5-дигидро-пиразол-1-ил)-4(1Н)-пиримидинона как двухосновной СН-кислоты и выявлено направление присоединения его моноаниона к 5-бензилиден-2,4,6(1Н,3Н,5Н)-пиримидинтриону в реакции Михаэля;
• рассмотрены причины образования продуктов электрофильного замещения в 5-гидрокси-3-метил-1-(6-метил-2-метилтиопиримидин-4-ил)пиразоле с исключительным участием пятичленного кольца субстрата;
• показана реакционная способность тиоамидной группы 1-(3,6-диметил-4-оксо-3,4-дигидропиримидин-2-ил)тиосемикарбазидов в синтезе тиазолов по Ганчу и установлена предпочтительная таутомерная форма существования возникающих при этом 2-(4-арилтиазол-2-ил)-1-(3,6-диметил-4-оксо-3,4-дигидропиримидин-2-ил)гидразинов.
Практическая значимость.
1. Разработан унифицированный способ получения 2-амино-5-бром-6-метил-4(3Н)-пиримидинонов, содержащих алкильные, циклоалкильные и аралкильные заместители у экзоциклического атома азота, бромированием соответствующих 2-амино-6-метил-4(3Н)-пиримидинонов in situ;
2. Предложен метод синтеза 2-гидразино-4-оксо-3,4-дигидроциклопента[d]пиримидина, который не требует использования 2-метилтиоциклопента[d]пиримидин-4(3Н)-она в качестве исходного субстрата;
3. Усовершенствован способ получения 6-метил-2-(3-метил-5-оксо-2,5-дигидропиразол-1-ил)-4(1Н)-пиримидинона, исключающий необходимость предварительного генерирования (6-метил-4-оксо-3,4-дигидропиримидин-2-ил)гидразона этилацетоацетата;
4. Выявлены сильные ингибиторы роста клеток микобактериальных (Mycobacterium smegmatis) и бактериальных (Stapylococcus aureus) культур в ряду 2-амино-4-галоген-ариламино-6-метилпиримидинов c варьируемыми заместителями в положении 2 гетерокольца и в ароматическом фрагменте и определены факторы, ответственные за проявление этими соединениями ингибирующих свойств.
5. Синтезированы 4-ариламинометилен-3-метил-1-(6-метил-4-оксо-3,4-дигидропирими-дин-2-ил)пиразол-5(4Н)-оны, которые обладают значительным ингибирующим действием в отношении микобактерий туберкулеза штамма H 37 Rv;
6. Осуществлена передача 2-бензиламино-4-(4-йодфенил)амино-6-метилпиримидина и наиболее активных его производных для расширенного предклинического тестирования, в том числе для определения величин острой и хронической токсичности полученных соединений.
Апробация работы. Результаты диссертационной работы доложены и обсуждены на:
конференции «Наукоемкие химические технологии». Волгоград, 2004;
VIII молодежной научной школе-конференции по органической химии. Казань, 2005;
XI международной конференции «Наукоемкие химические технологии». Самара, 2006;
научно-практической конференции, посвященной 182-й годовщине образования СПбГТИ (ТУ). Санкт-Петербург, 2010.
Публикации. По теме диссертации опубликована 21 работа, в том числе 17 статей в Журнале общей химии и тезисы 4 докладов на конференциях.
Объем и структура работы. Диссертационная работа изложена на 350 страницах и состоит из введения, аналитического обзора, обсуждения результатов, экспериментальной части и выводов. Работа содержит 36 таблиц и 33 рисунка. Список использованной литературы включает 244 источника.
ОСНОВНОЕ СОДЕРЖАНИЕ РАБОТЫ
Существенные различия в химическом поведении соединений двух обсуждаемых ниже групп соединений – амино- и гидразинопиримидинов – диктуют очевидную необходимость их раздельного рассмотрения. Вместе с тем, исходным субстратом для синтеза многих соединений обеих групп мы выбрали 6-метил-2-метилтио-4(3Н)-пиримидинон (1), поскольку именно он зарекомендовал себя как обеспечивающий наиболее селективный переход к амино- и гидразинопиримидинам.
1 Синтез и биологическая активность 2-амино-5-бром-6-метил-4(3Н)-пиримидинонов
С целью установления влияния замещения одного из протонов аминогруппы в 5-бром-6-метилизоцитозине, низкомолекулярном индукторе интерферона и противовирусном агенте, алкильным, циклоалкильным и аралкильным радикалами на биологические свойства этого соединения мы синтезировали 2-амино-5-бром-6-метил-4(3Н)-пирими-диноны (2 а-в) [R = Bu (a), цикло-C6H11 (б), PhCH2 (в)] через получение промежуточных 2-амино-6-метил-4(3Н)-пиримидинонов (3 а-в) аминированием метилтиоэфира (1) в соответствии с нижеследующей схемой:
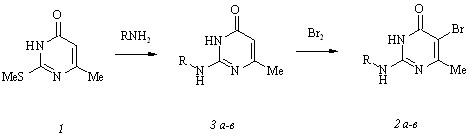
Аминирование соединения (1) бутил-, циклогексил- и бензиламинами протекает при 150-1600С в отсутствие растворителя и соотношении субстрат-реагент 1:3. Несмотря на имеющиеся в литературе утверждения, нам не удалось получить аминопиримидиноны (3 а-в) в кристаллическом виде прибавлением к реакционному остатку различных растворителей. Между тем, учитывая способность изоцитозинов образовывать устойчивые соли с минеральными кислотами, мы выделили соединения (3 а-в) в виде гидрохлоридов путем пропускания тока сухого хлороводорода через раствор реакционной смеси в абсолютном 2-пропаноле.
Сложности выделения аминопиримидинонов (3 а-в) в виде свободных оснований вызывают определенные затруднения при их бромировании, которые удается преодолеть, осуществляя обработку соединений (3 а-в) in situ бромом в ледяной уксусной кислоте. Образующиеся при этом гидробромиды 2-амино-5-бром-6-метил-4(3Н)-пиримидинонов (2 а-в) превращаются в свободные основания под действием водного раствора гидроксида натрия.
Изучение интерферон-индуцирующих свойств и противовирусной активности бром-аминопиримидинонов (2 а-в) по отношению к вирусу простого герпеса I типа (HSV-I) показало отсутствие таковых у синтезированных соединений в интервале концентраций 0-100 мкг/мл и позволило на основании этого факта сформулировать вывод о негативном влиянии замещения протона экзоциклической аминогруппы 5-бром-6-метилизоцитозина на рассматриваемые виды биологической активности.
2 Молекулярная структура 2-амино-6-метил-4(3Н)-пиримидинонов
Чтобы подтвердить или опровергнуть приписываемую аминопиримидинонам типа (3 а-в) структуру 4-оксо-3,4-дигидротаутомеров, мы изучили прототропную таутомерию 2-(2-гидроксиэтил)амино-6-метил-4-оксопиримидина (4) в нейтральном водном растворе методом УФ спектроскопии. Соединение (4) образуется при взаимодействии метилтиоэфира (1) с 2-кратным мольным избытком 2-аминоэтанола при 140-1500С в отсутствие растворителя. Выбор объекта исследования обусловлен возможностью получения пиримидиламиноэтанола (4) в виде свободного основания после нейтрализации избытка амина уксусной кислотой, малой вероятностью влияния концевой гидроксильной группы на таутомерные превращения изучаемого соединения, а также необходимостью последующего исследования его химических превращений, отличных от гетероциклизации.
Для получения модельных 2-(2-гидроксиэтил)амино-3,6-диметил-4(3Н)-пиримиди-нона (5) и 2-(2-гидроксиэтил)амино-1,6-диметил-4(1Н)-пиримидинона (6) нами использовано аминирование 3,6-диметил-2-метилтио-4(3Н)-пиримидинона (7) и 1,6-диметил-2-метилтио-4(1Н)-пиримидинона (8) соответственно 2-аминоэтанолом в вышеописанных условиях. Другое модельное соединение, 2-(2-гидроксиэтил)амино-6-метил-4-метокси-пиримидин, может быть синтезировано в виде О-ацетата (9) из пиримидиламиноэтанола (4) через промежуточные 2-(2-ацетоксиэтил)амино-6-метил-4-оксопиримидин (10) и 2-(2-ацетоксиэтил)амино-6-метил-4-хлорпиримидин (11):

Ацетилирование соединения (4) 1.5-кратным избытком ацетангидрида при 1000С (способ а) приводит к ацетату (10), который в результате обменного хлорирования оксо-трихлоридом фосфора превращается в хлорпиримидин (11). Последний, взаимодействуя с 3-кратным избытком метилата натрия в абсолютном метаноле, дает 2-(2-ацетоксиэтил)-амино-6-метил-4-метоксипиримидин (9). Деацетилирование соединения (10) действием гидроксида натрия в водном растворе не позволяет получить продукт расщепления сложноэфирной связи в кристаллическом виде по причине его высокой гигроскопичности.
Нейтральная форма. УФ спектры пиримидиламиноэтанола (4) обращают на себя внимание широкой асимметричной полосой поглощения с максимумом 272 нм (lg 3.71) и плечом при 285 нм в интервале концентраций (0.5-1.5)·10-4 моль/л. Вместе с тем оптическая плотность растворов соединения (4) зависит от концентрации линейно лишь диапазоне (0.5-1.25)·10-4 моль/л, а при более высоких концентрациях характеризуется отрицательным отклонением от закона Бугера. Это может свидетельствовать о гидратации пиримидиламиноэтанола (4), влекущей за собой его прототропную перегруппировку. Чтобы подтвердить факт гидратации соединения (4), мы сравнили его спектры в воде, этаноле и ДМФА и обнаружили сужение полосы поглощения и ее батохромный сдвиг до 289 нм (этанол) и 290 нм (ДМФА), сопровождающийся гиперхромным эффектом [lg 3.98 (этанол) и 4.00 (ДМФА)], при уменьшении полярности растворителя (значения нормализованного параметра полярности Димрота-Райхардта ETN для воды, этанола и ДМФА составляют соответственно 1.000, 0.654 и 0.404). Подобная отрицательная сольватохромия пиримидил-аминоэтанола (4) служит качественным критерием его гидратации.
Наличие в молекуле соединения (4) двух потенциальных центров гидратации – амидного и гуанидинового – требует оценки степени участия каждого из них в образовании водородных связей. При сопоставлении УФ спектров пиримидиламиноэтанола (4) при различных значениях рН со спектрами метиламинопиримидинона (5) и метоксипиримидина (9) видна близость спектров его нейтральной (рН 7) и анионной (рН 12) форм со спектром соединения (9), указывающая на предпочтительную гидратацию амидного фрагмента. В случае альтернативной гидратации гуанидинового фрагмента следовало ожидать подобия спектров нейтральной и катионной (рН 1) форм пиримидиламиноэтанола (4) спектру метиламинопиримидинона (6), которое не наблюдается в действительности.
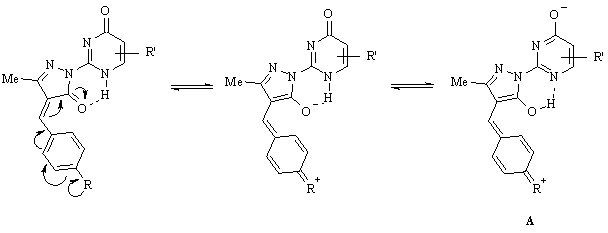
Несмотря на схожесть спектров соединений (4) и (9), полосу с максимумом 272 нм в спектре первого трудно связать с индивидуальным поглощением 4-гидрокситаутомера вследствие ее асимметричного характера. С другой стороны, отсутствие изосбестической точки в наборе кривых поглощения пиримидиламиноэтанола (4) при варьировании концентрации не позволяет судить о числе возможных таутомеров в его растворе. Для преодоления этих затруднений мы осуществили тесты на n-компонентность и пришли к выводу о том, что растворы соединения (4) не являются однокомпонентными. Оптическая плотность Di любого из них зависит от средней оптической плотности Dср всех растворов при любой длине волны линейно, однако необходимое условие прохождения этой зависимости через начало координат не выполняется. Напротив, постоянство отношения оптических плотностей двух растворов при любой длине волны в пределах погрешности дает право утверждать о присутствии в растворах пиримидиламиноэтанола (4) двух таутомеров. Из возможного числа таковых наиболее вероятными можно считать гидратированные 4-оксо-3,4-дигидро- (4 а) и 4-гидрокси- (4 б) таутомеры. Это подтверждается наличием изосбестической точки при 306 нм в спектрах соединения (4), метиламинопиримидинона (5) и метоксипиримидина (9). В дополнение к сказанному необходимо отметить, что, судя по имеющейся в спектрах нейтральной и анионной форм пиримидиламиноэтанола (4) и соединения (9) изосбестической точке при 287 нм, взаимные перегруппировки таутомеров (4 а) и (4 б) протекают через промежуточное образование цвиттериона А:
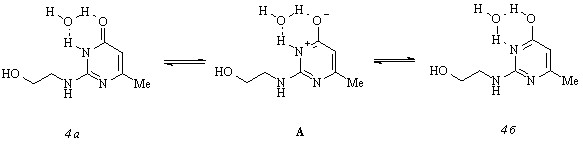
Неожиданная схожесть спектров соединения (4) в протонном этаноле и апротонном ДМФА заставляет предположить, что его прототропные перегруппировки обусловлены не только специфическим образованием водородных связей, но и высокой полярностью воды ( 78.30). Действительно, при добавлении менее полярного этанола ( 24.55) к водным растворам пиримидиламиноэтанола (4) происходит резкий батохромный сдвиг максимума полосы его поглощения, который достигает максимума полосы поглощения метиламинопиримидинона (5) при концентрации этанола выше 20% (об.). Иными словами, увеличение полярности смеси этанол-вода, количественно описываемой нормализованным параметром полярности Димрота-Райхардта ETN, вызывает гипсохромное смещение максимума полосы поглощения соединения (4). Эти данные убедительно показывают, что существование равновесной смеси таутомеров (4 а) и (4 б) возможно лишь в водных растворах с полярностью не ниже ETN 0.95. В растворах, характеризующихся полярностью в интервале ETN 0.95-0.92, имеет место переход 4-гидрокси- (4 б) в 4-оксо-3,4-дигидро- (4 а) таутомер, и исключительно в форме последнего пиримидиламиноэтанол (4) существует в растворах с полярностью менее ETN 0.92.
Протонированная форма. Независимо от типа таутомера аминопиримидинонов (3 а-в), существующего в водном растворе, протонирование его в кислых растворах приводит к образованию смеси катионов А и Б с преобладанием первого. Это следует из близости величин максимумов полос поглощения в УФ спектрах водных растворов гидрохлорида бутиламинопиримидинона (3 а) и метиламинопиримидинона (6), моделирующего катион А. Наличие в спектре соединения (3 а) плеча в области 290 нм, совпадающего по положению с максимумом полосы поглощения метиламинопиримидинона (5), указывает на присутствие в растворе некоторого количества катиона Б:

Причина предпочтительного образования катиона А в водном растворе заключается в его специфической сольватации с возникновением структуры В, стабилизированной водородными связями. Образование подобной структуры в случае катиона Б, очевидно, не возможно.
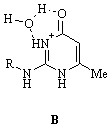
Аналогичный вывод о строении протонированных форм справедлив в отношении других членов ряда, гидрохлоридов циклогексил- и бензиламинопиримидинонов (3 б) и (3 в), однако при этом необходимо иметь в виду, что полоса поглощения, наблюдаемая в УФ спектре соединения (3 в), может представляться суперпозицией полос, обусловленных поглощением пиримидинового и фенильного колец. Подтверждением высказанному предположению служит заметно бльшая интенсивность полосы поглощения бензиламинопиримидинона (3 в) по сравнению с интенсивностями полос поглощения бутил- и циклогексиламинопиримидинонов (3 а) и (3 в).
3 Электрофильное замещение
в 2-(2-гидроксиэтил)амино-6-метил-4(3Н)-пиримидиноне
Пиримидиламиноэтанол (4) сочетает в себе свойства алифатического спирта и гетероцикла, активированного присутствием электронодонорных заместителей, и в этой связи представляет несомненный интерес как потенциальный объект атаки электрофильными реагентами.
Ацетилирование. Удовлетворительный (27%) выход ацетата (10), достигнутый способом а, побудил нас изыскать пути повышения выхода этого соединения посредством варьирования условий проведения реакции. Порционное прибавление ацетангидрида к суспензии пиримидиламиноэтанола (4) в абсолютном пиридине, нагретой до 800С, и последующая кристаллизация продукта (способ б) позволили увеличить выход ацетата (10) до 55%. Той же величины выхода соединения (10) мы достигли, обрабатывая суспензию пиримидил-аминоэтанола (4) ацетилхлоридом в абсолютном пиридине при 0-50С (способ в).
Карбамоилирование. Учитывая то обстоятельство, что карбамоилирование пиримидинов, содержащих экзоциклические гидрокси- и аминогруппы одновременно, может протекать по обоим реакционным центрам, мы изучили взаимодействие пиримидиламиноэтанола (4) с фенилизоцианатом. При нагревании эквимольных количеств исходных компонентов в кипящем абсолютном пиридине (способ а) нами получен 6-метил-2-[2-(фенилкарбамоил-окси)этил]амино-4(3Н)-пиримидинон (12) с выходом 18%, считая на очищенный кристаллизацией продукт. Замена пиридина абсолютным ДМФА и проведение реакции при 1200С (способ б) позволяет увеличить выход фенилкарбамата (12) до 50-55%. Имеющее место увеличение выхода соединения (12) можно связать с более эффективным разделением атомных зарядов в гидроксильной группе благодаря проведению реакции в биполярном ДМФА, обладающем свойством диссоциирующего растворителя.
Для подтверждения структуры фенилкарбамата (12) химическим путем мы подвергли это соединение кислотному гидролизу концентрированной хлороводородной кислотой. Исследование состава гидролизата посредством хроматографии в присутствии соединений-свидетелей показало, что он не содержит ни анилина, ни пиримидиламиноэтанола (4), ни 6-метилурацила. Более того, молярные массы продукта гидролиза и фенилкарбамата (12) оказались равны. В сочетании с совокупностью спектральных данных это указывает на внутримолекулярную перегруппировку соединения (12) в реализованных условиях в 2-(2-гидроксиэтил)амино-6-метил-5-фенилкарбамоил-4(3Н)-пиримидинон (13).

Прямое галогенирование. Наличие в структуре соединения (4) ряда электронодонорных заместителей позволяет рассчитывать на успешное его галогенирование молекулярными бромом и йодом. 5-Бром-2-(2-гидроксиэтил)амино-6-метил-4(3Н)-пиримидинон (14) мы получили действием брома на исходный субстрат (4) в растворе ледяной уксусной кислоты при 250С с выходом около 80%:

Cхожим образом нами проведено йодирование пиримидиламиноэтанола (4) кристаллическим йодом в 10%-ном водном растворе гидроксида натрия при 250С с получением 2-(2-гидроксиэтил)амино-5-йод-6-метил-4(3Н)-пиримидинона (15), выход которого составил около 35%.
Метилирование. Для выявления приоритетных направлений алкилирования соединений типа (3 а-в) с участием атомов амбидентной системы мы исследовали метилирование пиримидиламиноэтанола (4) в различных средах при фиксированных времени контакта реагентов (120 ч) и температуре (25±50С).
Метилирование соединения (4) диметилсульфатом в водном растворе гидроксида натрия дает метиламинопиримидинон (5), идентичный образцу того же соединения, полученному аминированием метилтиоэфира (7) 2-аминоэтанолом:

Образование соединения (5) в водно-щелочной среде с рН 12 обусловливается ионизацией амидного фрагмента пиримидиламиноэтанола (4), протекающей более чем на 99% и ведущей к образованию аниона А, в котором избыточный заряд сосредоточен на атоме кислорода. О локализации заряда на атоме кислорода свидетельствуют подобие УФ спектров соединения (4) при рН 12 и метоксипиримидина (9) и батохромный сдвиг максимума полосы поглощения аниона А до длины волны 289 нм, которая соответствует поглощению метиламинопиримидинона (5) в водно-щелочных средах соединения (4) при увеличении процентного содержания в них апротонного ацетонитрила. Наблюдаемый батохромный сдвиг и сопровождающий его гипохромный эффект указывают также на гидратацию анионного центра пиримидиламиноэтанола (4) растворителем. Это вызывает блокирование ионизированного атома кислорода водородными связями и тем самым обеспечивает протекание метилирования исходного субстрата по атому N3.
Образованием метиламинопиримидинона (5) характеризуется также метилирование соединения (4) метилйодидом в этаноле в присутствии гидроксида калия при 250С, однако высокая селективность взаимодействия в данном случае вызвана блокированием атома кислорода катионом металла. Сказанное подтверждается совпадением УФ спектров пиримидиламиноэтанола (4) в этаноле в отсутствие и в присутствии основания и наличием точки перегиба зависимости оптической плотности этанольных растворов соединения (4) от мольного соотношения субстрат-гидроксид калия. Проецирование точки перегиба на ось абсцисс свидетельствует об образовании комплекса Б между пиримидиламиноэтанолом (4) и гидроксидом калия состава 1:1.

Резкое снижение селективности метилирования соединения (4) происходит при замене этанола ДМФА и гидроксида калия карбонатом калия. Несмотря на близость величин диэлектрической проницаемости этанола ( 24.6) и ДМФА ( 36.71), блокирования атома кислорода основанием, как в предыдущем случае, не происходит по причине сольватации катиона калия растворителем, и в продуктах реакции присутствует смесь метил-аминопиримидинона (5) и 2-(2-гидроксиэтил)амино-6-метил-4-метоксипиримидина (16). Сложность выделения последнего в виде свободного основания не позволяет определить соотношение образующихся изомеров и оценить таким образом реакционную способность амбидентного центра соединения (4) по отношению к метилирующему агенту в реализованных условиях. Строение метоксипиримидиламиноэтанола (16), выделенного из реакционной массы в виде пикрата, доказано нами встречным щелочным гидролизом соединения (9) гидроксидом натрия в водном растворе и последующей кватернизацией свободного основания пикриновой кислотой.
4 Синтез и биологическая активность
4-ариламино-2-(2-ацетоксиэтил)амино-6-метилпиримидинов
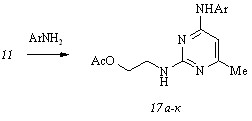
Для получения неописанных 4-ариламино-2-(2-ацетоксиэтил)амино-6-метилпирими-динов (17 а-к) и последующей оценки их биологического действия по отношению к имеющимся биологическим объектам мы изучили конденсацию хлорпиримидина (11) с рядом ароматических аминов.
Удовлетворительная величина выхода хлорпиримидина (11) и сложность достижения ее постоянства при обменном хлорировании ацетата (10) заставили нас обратиться к изучению взаимодействия последнего с пентахлоридом фосфора. Обработкой ацетата (10) пентахлоридом фосфора при 100-1100С мы получили хлорпиримидин (11) с выходом 50%, считая на очищенный кристаллизацией продукт. При последующих воспроизведениях данного способа обменного хлорирования ацетата (10) выход соединения (11) колебался в пределах ±5% от указанного значения.
Конденсация хлорпиримидина (11) с анилинами протекает в отсутствие растворителя при температуре 1200С и мольном соотношении реагентов 1:1 или 1:2. Отказ от проведения взаимодействия в растворителе и применение повышенной температуры вызвано пониженной подвижностью атома галогена в соединениях, структурно схожих с субстратом (11). После обработки продукта реакции хлороводородной кислотой мы выделяли арил-аминопиримидины (17 а-к) в виде гидрохлоридов.
Извлечение ариламинопиримидинов (17 а-к) из реакционной массы в виде гидрохлоридов требует исследования направления их протонирования. Кватернизация простых 2,4-диаминопиримидинов происходит с участием кольцевого атома N1. Для подтверждения справедливости этого утверждения применительно к соединениям (17 а-к) мы сопоставили УФ спектры водных растворов гидрохлорида 2-(2-ацетоксиэтил)амино-6-метил-4-(4-метилфенил)аминопиримидина (17 в) и 6-метил-4-(4-метилфенил)амино-2(1Н)-пирими-динона (18) и, обнаружив их подобие в длинноволновой области, получили экспериментальное свидетельство о протонировании соединения (17 в) по атому N1. Получение арилцитозина (18) осуществлено нами аминированием 6-метил-4-метилтио-2(1Н)-пирими-динона (19) 4-метилфениламином в жестких условиях – при 1400С в отсутствие растворителя. Необходимый для этого метилтиопиримидинон (19) синтезирован S-метили-рованием 6-метил-4-тиоксо-3,4-дигидро-2(1Н)-пиримидинона (20) метилйодидом в водном растворе гидроксида натрия, а тиоурацил (20), в свою очередь, – сульфурированием 6-метилурацила (21) пентасульфидом фосфора в кипящем пиридине:
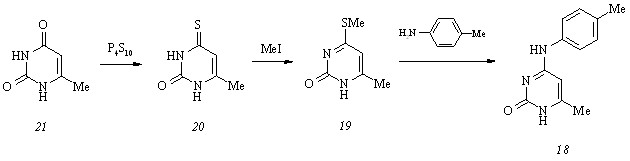
Биологический скрининг ариламинопиримидинов (17 а-к), проведенный с использованием бактериальных (Esherichia coli, Staphylococcus aureus), грибковых (Candida albicans, Aspergillus niger) и микобактериальных (Mycobacterium smegmatis) культур in vitro, показывает, что некоторые из них проявляют слабую антибактериальную активность по отношению к клеткам Staphylococcus aureus, большинство же обладает выраженным антимикобактериальным действием в интервале концентраций 12.5-100 мкг/мл (табл. 1).
Таблица 1 – Количественная оценка биологической активности
4-ариламино-2-(2-ацетоксиэтил)амино-6-метилпиримидинов (17 а-к)
| № соед. | Ar | Минимальная концентрация, обеспечивающая ингибирование биообъекта на 100% IC100, мкг/мл | ||
| Esherichia coli | Staphylococcus aureus | Mycobacterium smegmatis | ||
| 17 а | Ph | - | - | - |
| 17 б | 3-MeC6H4 | - | - | 50 |
| 17 в | 4-MeC6H4 | - | - | 100 |
| 17 г | 4-MeOC6H4 | - | - | - |
| 17 д | 4-FC6H4 | 100 | 100 | 50 |
| 17 е | 3-ClC6H4 | - | 100 | 50 |
| 17 ж | 4-ClC6H4 | - | - | 50 |
| 17 з | 3-BrC6H4 | - | - | 12.5 |
| 17 и | 4-BrC6H4 | - | - | - |
| 17 к | 4-IC6H4 | - | 50 | 12.5 |
Наиболее активными оказались соединения (17 з) и (17 к), ингибировавшие размножение микроорганизмов Mycobacterium smegmatis на 100% в концентрации 12.5 мкг/мл. Учитывая то обстоятельство, что хлорпиримидин (11) и 2-(2-ацетоксиэтил)амино-4-цикло-гексиламино-6-метилпиримидин (22) не проявляют антимикобактериальных свойств, в первом приближении можно считать, что активность ариламинопиримидинов (17 а-к) по отношению к указанной культуре клеток обусловлена специфическими фармакофорными свойствами ариламинопиримидинового фрагмента их молекул.
5 Направленный поиск биологически активных агентов
в ряду 2-амино-4-ариламино-6-метилпиримидинов
В свете установленного факта увеличения уровня антимикобактериальной активности ариламинопиримидинов (17 а-к) с ростом параметра их липофильности lg P мы синтезировали 4-ариламино-6-метил-2-циклогексиламинопиримидины (23 а, б) и 4-ариламино-2-бензиламино-6-метилпиримидины (24 а, б), предполагая, что введение гидрофобного заметителя в их молекулы приведет к уменьшению концентрации ингибирования культуры клеток Mycobacterium smegmatis.
Получение гидрохлоридов циклогексиламинопиримидинов (23 а, б) и бензиламинопиримидинов (24 а, б) осуществлено нами аминированием 6-метил-4-хлор-2-циклогек-силаминопиримидина (25) и 2-бензиламино-6-метил-4-хлорпиримидина (26) соответственно 3-бром- и 4-йодфениламинами в отсутствие растворителя при температурах 80-1200С и мольном соотношении реагентов 1:1. Аминохлорпиримидины (25) и (26) мы синтезировали обменным хлорированием аминопиримидинонов (3 б, в) оксотрихлоридом фосфора.
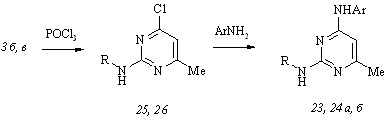
R = цикло-C6H11 (a), PhCH2 (б); Ar = 3-BrC6H4 (а), 4-IC6H4 (б)
Выдвинутая гипотеза подтверждается лишь частично. Бромзамещенный циклогек-силаминопиримидин (23 а) показал близкий по величине эффект ингибирования рассматриваемого биобъекта, в то время как бромфенилбензиламинопиримидин (24 а) оказался полностью неактивным в интервале концентраций 0-100 мкг/мл по сравнению с соединением-прототипом (17 з). Активность йодфенилциклогексиламинопиримидина (23 б) снизилась приблизительно в 4 раза, а антимикобактериальное действие йодзамещенного бензиламинопиримидина (24 б) возросло более чем в 7 раз в сопоставлении с препаратом (17 к). Кроме того, соединения (23 б, 24 а) и особенно йодфенилбензиламинопиримидин (24 б) проявили сильную ингибирующую способность по отношению к клеточной культуре Staphylococcus aureus (табл. 2).
Результаты биологического тестирования 2-амино-4-(3-бромфенил)амино-6-метил-пиримидинов (17 з, 23 а и 24 а) свидетельствуют о том, что активность этих соединений обусловлена не только природой радикала, находящегося у атома N2, но и местом расположения галоидного заместителя. Для установления влияния перемещения атома брома из ароматического кольца бромфениламинопиримидинов (17 з, 23 а и 24 а) в гетероядро на их ингибирующую способность по отношению к клеткам культур Mycobacterium smegmatis и Staphylococcus aureus мы синтезировали их изостерические аналоги, гидрохлориды 2-амино-5-бром-6-метил-4-фениламинопиримидинов (27 а-в), и оценили биологические свойства полученных соединений.
Таблица 2 – Количественная оценка биологической активности
4-ариламино-6-метил-2-циклогексиламинопиримидинов (23 а, б)
и 4-ариламино-2-бензиламино-6-метилпиримидинов (24 а, б)
| № соед. | Минимальная концентрация, обеспечивающая ингибирование биообъекта на 100% IC100, мкг/мл | |
| Mycobacterium smegmatis | Staphylococcus aureus | |
| 23 а | 10 | 100 |
| 23 б | 50 | 25 |
| 24 а | - | 25 |
| 24 б | 1.7 | 6.2 |
2-(2-Ацетоксиэтил)амино-5-бром-6-метил-4-фениламинопиримидин (27 а) получен нами аминированием 2-(2-ацетоксиэтил)амино-5-бром-6-метил-4-хлорпиримидина (28) эквимольным количеством фениламина в отсутствие растворителя при 100-1100С, а ключевой интермедиат (28) – химическими трансформациями бромпиримидинона (14), включающими его ацетилирование ацетангидридом в пиридине при 1000С и обменное хлорирование 2-(2-ацетоксиэтил)амино-5-бром-6-метил-4(3Н)-пиримидинона (29), или альтернативным бромированием хлорпиримидина (11):
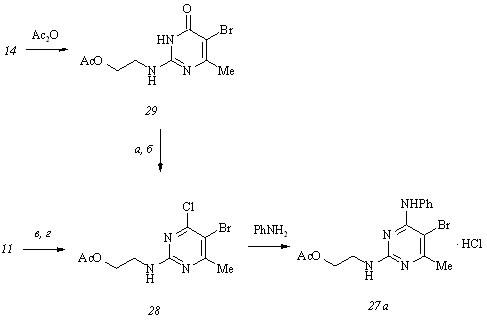
Обменное хлорирование бромпиримидина (29) позволяет получать дигалогенпиримидин (28) лишь с низкими (3-10%) выходами независимо от типа используемого хлорирующего агента [оксотрихлорид фосфора (способ а) и пентахлорид фосфора (способ б)]. Бльших величин (до 40%) выхода дигалогенпиримидина (28) удается достичь непосредственным бромированием хлорпиримидина (11) бромом в присутствии акцептора бромоводорода, триэтиламина (способ в), и особенно N-бромсукцинимидом (NBS) (способ г) в тетрахлорметане.
Синтез гидрохлорида 5-бром-6-метил-4-фениламино-2-циклогексиламинопирими-дина (27 б) мы провели, исходя из циклогексиламинохлорпиримидина (25), который подвергали аминированию эквимольным количеством фениламина в отсутствие растворителя при 100-1100С. Полученный гидрохлорид 6-метил-4-фениламино-2-циклогексиламино-пиримидина (30) обрабатывали избытком гидроксида натрия в воде и выделяли свободное основание (31), которое при взаимодействии с бромом в присутствии триэтиламина (способ а) или с NBS (способ б) в тетрахлорметане превращалось в 5-бром-6-метил-4-фенил-амино-2-циклогексиламинопиримидин (32). В заключение образовавшееся свободное основание (32) действием хлороводородной кислоты переводили в целевой гидрохлорид (27 б).


Для получения гидрохлорида 2-бензиламино-5-бром-6-метил-4-фениламинопири-мидина (27 в) мы реализовали схожую с описанной последовательность трансформаций бензиламинохлорпиримидина (26). Аминирование соединения (26) эквимольным количеством фениламина в отсутствие растворителя при 100-1100С приводит к гидрохлориду 2-бензиламино-6-метил-4-фениламинопиримидина (33), нейтрализация которого избытком гидроксида натрия в воде дает свободное основание (34). Последнее при обработке NBS превращается в 2-бензиламино-5-бром-6-метил-4-фениламинопиримидин (35). Полученное соединение (35) после суспендирования в хлороводородной кислоте выделяется в виде гидрохлорида бензиламинобромпиримидина (27 в).
Перемещение атома галогена из ароматического кольца гидрохлоридов ариламинопиримидина (17 з) и бромфениламинопиримидина (23 а) в гетероциклическое ядро вызывает резкое уменьшение ингибирующего действия возникающих при этом гидрохлоридов 5-бром-2,4-диаминопиримидинов (27 а-в). Гидрохлорид бензиламинобромпиримидина (27 в), как и гидрохлорид его изостерического аналога, бромфенилбензиламинопиримидина (24 а), не обладает антимикобактериальной активностью в интервале концентраций 0-100 мкг/мл, но подавляет рост клеток культуры Staphylococcus aureus на 100% в концентрации 50 мкг/мл. В целом, полученные данные позволяют утверждать об утрате 5-бром-2,4-диаминопиримидинами (27 а-в), содержащими атом брома в гетерокольце, фармакофорных свойств по отношению к клеткам Mycobacterium smegmatis.
6 Пути модификации структуры
2-бензиламино-4-(4-йодфенил)амино-6-метилпиримидина
Совокупный анализ данных биологической активности ариламинопиримидинов (17 а-к), циклогексил- и бензиламинопиримидинов (23 а, б) и (24 а, б), а также 5-бром-2,4-диаминопиримидинов (27 а-в) позволяет признать справедливость первоначальной гипотезы о фармакофорной природе ариламинопиримидинового фрагмента рассматриваемых соединений и вынуждает отказаться от предположения о симбатности изменения их свойств – уровня биологической активности и параметра липофильности. Последнее имеет место лишь в ряду соединений (17 а-к) с варьируемым заместителем в фенильном кольце. Более того, наблюдаемое увеличение способности к ингибированию роста клеток отдельных биологических объектов при переходе от ариламинопиримидина (17 к) к бензиламинопиримидину (24 б) не обладает, по всей видимости, характером закономерности и инспирирует таким образом анализ элементов структуры соединения (24 б), вносящих определенный вклад в его биологическое действие. Для выявления означенных элементов мы предприняли синтез аналогов бензиламинопиримидина (24 б) оценили их ингибирующие свойства по отношению к клеточным культурам Mycobacterium smegmatis и Staphylococcus aureus.
В основу синтеза гидрохлоридов всех соединений (36, а-ж) положена унифицированная схема, включающая последовательные аминирование тиоэфира (1), обменное хлорирование 2-амино-6-метил-4(3Н)-пиримидинонов (3 г-к) оксотрихлоридом фосфора и заключительное аминирование 2-амино-6-метил-4-хлорпиримидинов (37 а-ж) эквимольными количествами анилинов в отсутствие растворителя при 95-1300С.
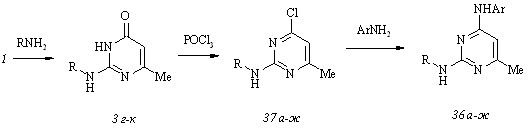
2-Бензилтио-4-(4-йодфенил)амино-6-метилпиримидин (38) получен нами аминированием 2-бензилтио-6-метил-4-хлорпиримидина (39) 4-йодфениламином, а необходимый для этого тиохлорпиримидин (39) синтезирован обменным хлорированием 2-бензилтио-6-метил-4(3Н)-пиримидинона (40) оксотрихлоридом фосфора.
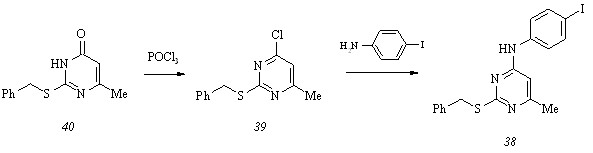
Количественные данные процессов ингибирования клеток Mycobacterium smegmatis и Staphylococcus aureus (табл. 3) дают возможность сформулировать общие выводы относительно связи типа «структура-активность» в ряду аналогов бензиламинопиримидина (24 б):
1. высоким уровнем биологической активности обладают 4-йодфениламинопири-мидины, характеризующиеся минимальной степенью модификации структуры по сравнению с соединением-прототипом (24 б);
2. увеличение количества метиленовых групп в заместителе у атома N4 приводит к изменению вида биологического действия соединения (24 б).
Ингибирование роста микобактерий. Антимикобактериальные свойства присущи 2-амино-4-(йодфенил)минопиримидинам (36 а-ж), имеющим в своей структуре алкил-, циклоалкил- или аралкиламиногруппу в положении 2.
Таблица 3 – Количественная оценка биологической активности
2-амино-4-(йодфенил)амино-6-метилпиримидинов (36 а-ж)
| № соед. | R | Ar | Минимальная концентрация, обеспечивающая ингибирование биообъекта на 100% IC100, мкг/мл | |
| Mycobacterium smegmatis | Staphylococcus aureus | |||
| 36 а | Ph(CH2)2 | 4-IC6H4 | 25 | 5 |
| 36 б | Ph(Me)CH | 4-IC6H4 | 2.5 | - |
| 36 в | С4H3OCH2a | 4-IC6H4 | - | - |
| 36 г | 4-MeC6H4 | 4-IC6H4 | 25 | - |
| 36 д | PhCH2 | 3-IC6H4 | 2.5 | - |
| 36 е | Hб | 4-IC6H4 | - | 100 |
| 36 ж | PhNHC(O)O(CH2)2 | 4-IC6H4 | - | 5 |
Примечание: а. (Фуран-2-ил)метил; б. Ингибирует рост клеток Mycobacterium tuberculosis, IC100 12.5 мкг/мл
Ингибирование роста бактерий. Упомянутая способность соединений (17 к), (23 б), (24 б) и (36 а, ж) ингибировать рост клеток Staphylococcus aureus заставляет подвергнуть анализу количественные характеристики этого процесса. Симбатность изменения меры антибактериальной активности, выражаемой логарифмом обратной молярной концентрации lg 1/С0, и поляризуемости Р соединений (17 к), (23 б), (24 б), (36 а) и (36 ж) (табл. 4) указывает на существование корреляции между указанными параметрами.
Таблица 4 – Зависимость антибактериальной активности
4-йодфениламинопиримидинов (17 к), (23 б), (24 б), (36 а) и (36 ж)
от поляризуемости их молекул
| № соед. | Р, | lg 1/C0 |
| 17 к | 34.48 | 3.91 |
| 23 б | 33.80 | 4.21 |
| 24 б | 35.63 | 4.82 |
| 36 а | 37.47 | 4.93 |
| 36 ж | 41.38 | 4.99 |
Конкретизация вида корреляционной зависимости lg 1/C0 = f (P) (экспоненциальная или параболическая) возможна лишь при получении минимум одного значения логарифма обратной молярной концентрации аналога 4-йодфениламинопиримидинов (17 к), (23 б), (24 б), (36 а) и (36 ж) с величиной поляризуемости, превышающей наибольшую из указанных. Для этой цели мы синтезировали гидрохлорид 4-(4-йодфенил)амино-6-метил-2-[2-(4-этилфенокси)этил]аминопиримидина (41), который имел расчетное значение Р, равное 42.23, и в ходе биологического тестирования определили соответствующую этому соединению величину lg 1/C0.

Получение гидрохлорида йодфенилфеноксиэтиламинопиримидина (41) осуществлено нами по схеме, использованной для синтеза соединений (36 а-ж), исходя из фенилкарбамата (12). Рассчитанная на основании результатов биологического скрининга соединения (41) величина lg 1/C0 = 4.31 позволяет осуществить выбор вида корреляции lg 1/C0 = f (P) в пользу параболической зависимости (см. рис.):

Рисунок – Антибактериальная активность 4-йодфениламинопиримидинов
(17 к), (23 б), (24 б), (36 а), (36 ж) и (41) как функция поляризуемости их молекул
Несмотря на невысокий (r 0.83) коэффициент корреляции, ценность получения данной зависимости заключается в возможности постулирования максимального или близкого к таковому уровня антибактериальной активности аналогов 4-йодфениламинопирими-динов (17 к), (23 б), (24 б), (36 а), (36 ж) и (41), характеризующихся параметром поляризуемости в интервале 38-40.
7 Структуры потенциальных метаболитов
2-бензиламино-4-(4-йодфенил)амино-6-метилпиримидина
Дополнительную информацию к установлению факторов, ответственных за проявление бензиламинопиримидином (24 б) антимикобактериальной активности, может предоставить исследование структуры продуктов, образующихся в процессе метаболизма этого соединения. Следует отметить, что гидролиз бензиламинопиримидина (24 б) с участием обеих экзоциклических связей C-N представляется маловероятным путем его начальной биотрансформации, так как расщепление связей С2-N и С4-N под действием воды теоретически приводит либо к 4-(4-йодфенил)амино-6-метил-2(1Н)-пиримидинону (42), либо к бензиламинопиримидинону (3 в) соответственно. В первом случае активность всех 4-йодфениламинопиримидинов (36 а-ж) обусловлена метаболическим переходом в йодфенилцитозин (42) и, очевидно, не зависит от природы заместителя у атома N2, что находится в явном противоречии с количественными данными об их биологической активности. С другой стороны, наличие у соединений (36 г-з) антимикобактериальных свойств едва ли связано с их биотрансформацией в бензиламинопиримидинон (3 в) ввиду того, что последний полностью лишен способности к ингибированию роста клеток культуры Mycobacterium smegmatis, по крайней мере, в рассматриваемом диапазоне концентраций. Принимая во внимание сказанное выше, мы предположили, что в качестве потенциальных метаболитов бензиламинопиримидина (24 б) могут выступать соединение (33) и 2-бензиламино-4-(4-гидроксифенил)амино-6-метилпиримидин (43) как продукты первичной биотрансформации, выражающейся в восстановительном или гидролитическом дейодировании субстрата. Для проверки выдвинутой гипотезы нами реализован синтез гидроксифениламинопиримидина (43) и произведена количественная оценка антимикобактериальной активности соединений (33) и (43).
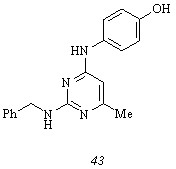
Получение гидроксифениламинопиримидина (43) осуществлено аминированием бензиламинохлорпиримидина (26) эквимольным количеством 4-аминофенола в отсутствие растворителя при температуре 1300С.
Результаты биологического скрининга синтезированного ранее фениламинопиримидина (33) и гидроксифениламинопиримидина (43) свидетельствуют о наличии способности к угнетению роста клеток Mycobacterium smegmatis у обоих соединений. Фениламинопиримидин (33) характеризуется концентрацией ингибирования IC100 25 мкг/мл, в то время как аналогичная величина для гидроксифениламинопиримидина (43) многократно уменьшается и составляет 5 мкг/мл. Приведенные данные указывают на предпочтительное метаболическое превращение бензиламинопиримидина (24 б) в соединение (43) путем ферментативного гидролиза.
8 Синтез и биологическая активность 4-арилиден-1-(4-оксо-3,4-дигидро-
пиримидин-2-ил)пиразол-5(4Н)-онов
Формальная замена углеводородного радикала в аминопиримидинонах (3 а-в) аминогруппой приводит к 2-гидразино-6-метил-4(3Н)-пиримидинону (44), предшественнику противовоспалительного препарата «Мепиризол» [6-метил-2-(3-метил-5-метоксипиразол-1-ил)-4-метоксипиримидин]. Для получения его аналогов мы синтезировали 4-арилиден-3-метил-1-(6-метил-4-оксо-3,4-дигидропиримидин-2-ил)пиразол-5(4Н)-оны (45 а-ж) в соответствии со схемой:

R = 4-Me2NC6H4 (a), 4-HOC6H4 (б), 4-Et2NC6H4 (в), 3-MeO-4-HOC6H3 (г), 3-EtO-4-HOC6H3 (д),
3-MeO-4-HO-5-СlC6H2 (е), 3-MeO-4-HO-5-BrC6H2 (ж)
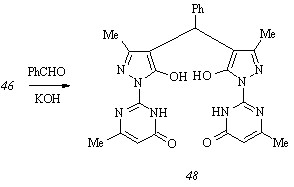
Конденсация гидразинопиримидинона (44) с этилацетоацетатом в отсутствие растворителя при 1000С дает (6-метил-4-оксо-3,4-дигидропиримидин-2-ил)гидразон этилацетоацетата (46), который, взаимодействуя с ароматическими альдегидами в этаноле в присутствии гидроксида калия, образует целевые арилиденпиразолоны (45 а-ж) (способ а). Существенным отличием представленной схемы от общепринятой является отсутствие необходимости выделения промежуточного 6-метил-2-(3-метил-5-оксо-4,5-дигидропиразол-1-ил)пиримидин-4(3Н)-она (47).
Особенность циклоконденсации пиримидилгидразона (46) с ароматическими альдегидами заключается в образовании ожидаемых соединений (45 а-ж) лишь в том случае, когда заместитель в альдегиде располагается в пара-положении кольца и обладает ауксохромным эффектом. При использовании альдегидов другого строения арилиденпиразолоны (45 а-ж) либо не возникают вовсе, либо присоединяют вторую молекулу пиримидилпиразолона (47) in situ, давая арилбис[5-гидрокси-3-метил-1-(6-метил-4-оксо-3,4-дигидро-пиримидин-2-ил)пиразол-4-ил]метаны, например соединение (48):
Предложенная схема синтеза арилиденпиразолонов (45 а-ж) непосредственно из пиримидилгидразона (46) и ароматических альдегидов с успехом применена нами при получении 2-(4-арилиден-3-метил-5-оксо-4,5-дигидропиразол-1-ил)циклопента[d]пиримидин-4(3Н)-онов (49 а, б):

R = 4-Me2NC6H4 (a), 4-Et2NC6H4 (б)
Для синтеза исходного субстрата этой реакционной последовательности, 2-гидр-азиноциклопента[d]пиримидин-4(3Н)-она (50), мы применили модифицированный способ, состоящий в циклоконденсации бензилиденаминогуанидина с этил-2-оксоциклопента-ноатом и последующем кислотном гидролизе образующегося 2-бензилиденгидразино-циклопента[d]пиримидин-4(3Н)-она. Данный способ позволяет избежать трудностей, связанных с рециклизацией 2-метилтиоциклопента[d]пиримидин-4(3Н)-она в ходе его гидразинолиза.
Роль ауксохромного заместителя соединений (45 а-ж) и (49 а, б) заключается в стабилизации их структуры посредством образованием резонансной формы А, обладающей лабильной внутримолекулярной водородной связью между псевдоионизированной карбонильной группой пиразольного кольца и группой N1H пиримидинового цикла:
В попытке получить гетероароматические аналоги арилиденпиразолонов (45 а-ж) нами обнаружены особенности циклоконденсации пиримидилгидразона (46) с некоторыми альдегидами ряда индола. При использовании незамещенного индол-3-карбоксальде-гида происходит образование ожидаемого 4-(индол-3-ил)метилен-3-метил-1-(6-метил-4-оксо-3,4-дигидропиримидин-2-ил)пиразол-5(4Н)-она (51), в то время как введение в реакцию его 2-метилпроизводного сопровождается выделением 3-метил-1-(6-метил-4-оксо-3,4-дигидропиримидин-2-ил)-4-[5-гидрокси-3-метил-1-(6-метил-4-оксо-3,4-дигидропири-мидин-2-ил)пиразол-4-ил]метиленпиразол-5(4Н)-она (52):
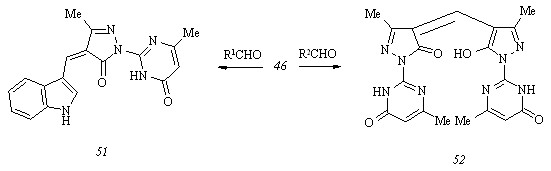
R1 = индол-3-ил; R2 = 2-метилиндол-3-ил
Биологическое тестирование арилиденпиразолонов (45 а-ж) и их аналогов, содержащих аннелированный циклопентановый фрагмент (49 а, б), проведенное с использованием бактериальных (Esherichia coli, Staphylococcus aureus), грибковых (Candida albicans, Aspergillus nieger) и микобактериальных (Mycobacterium tuberculosis) культур in vitro, показывает, что соединения (45 б) и (45 в) ингибируют рост клеток Mycobacterium tuberculosis в концентрациях 100 и 50 мкг/мл соответственно. Остальные соединения, в том числе бициклические (49 а, б), не обладают противотуберкулезными свойствами.
9 Особенности циклизации (3,6-диметил-4-оксо-3,4-дигидро-
пиримидин-2-ил)гидразона этилацетоацетата
В отличие от пиримидилгидразона (46) (3,6-диметил-4-оксо-3,4-дигидропиримидин-2-ил)гидразон этилацетоацетата (53), синтезированный нами конденсацией 2-гидразино-3,6-диметил-4(3Н)-пиримидинона (54) с этилацетоацетатом, взаимодействуя с 4-диметил-амино- и -диэтиламинобензальдегидами в вышеописанных условиях дает не подлежащие идентификации смолообразные продукты. Безуспешным оказывается также попытка получения соединений типа (45 а-ж) через стадию генерирования промежуточного 3,6-диметил-2-(3-метил-5-оксо-4,5-дигидропиразол-1-ил)-4(3Н)-пиримидинона вследствие отсутствия у N-метилпиримидилгидразона (53) тенденции к замыканию пиразольного цикла. В ходе нагревания с гидроксидом калия в этаноле при 500С соединение (53) образует смолоподобное вещество, а с более мягким деэтерифицирующим агентом, карбонатом калия, в разбавленном этаноле при той же температуре – неидентифицирумый высокоплавкий продукт. N-Метилпиримидилгидразон (53) выделяется неизменным после длительного кипячения в толуоле, но претерпевает неожиданную трансформацию при нагревании до 170-1900С. После окончания выдержки соединения (53) при указанных температурах мы выделили продукт, структурно соответствовавший 4,7-диметил-1,2,4-три-азоло[1,5-a]пиримидин-5(4Н)-ону (55):

Механизм обнаруженной аномальной циклизации N-метилпиримидилгидразона (53) включает специфическое расщепление его алифатического фрагмента с выделением этанола и генерированием неустойчивого бутенона А, который, элиминируя метилкетен, превращается в бицикл (55):

Элиминирование метилкетена от промежуточного бутенона А вызывает образование электронодефицитного карбена Б, способного к стабилизации структуры посредством циклизации либо в 5,8-диметил-1,2,4-триазоло[4,3-a]пиримидин-7(8Н)-он (56), либо в бицикл (55) с предварительным перераспределением электронной плотности и разрывом связи N-N. Первый маршрут может быть исключен из рассмотрения на основании отсутствия соединения (56) в продуктах термолиза N-метилпиримидилгидразона (53), а также ввиду его неподверженности термической изомеризации в бицикл (55). Синтез соединения (56) мы осуществили циклоконденсацией N-метилгидразинопиримидинона (54) с триэтилортоформиатом в отсутствие растворителя. Это взаимодействие позволяет получать преимущественно целевой продукт (56), а доля изомерного бицикла (55) в реакционной смеси не превышает 10%. Аналогичная степень региоселективности присуща реакции N-метилгидразинопиримидинона (54) с другими источниками одноуглеродных фрагментов – муравьиной кислотой и этилформиатом.
Причина стабилизации карбена Б замыканием триазоло[1,5-a]пиримидиновой системы (55) заключается в отсутствии влияния на этот процесс обратимой сольватации. Полагая, что продукт дегидратации 3,6-диметил-2-(2-формилгидразино)пиримидин-4(3Н)-она, возникающего первоначально из N-метилгидразинопиримидинона (54) и муравьиной кислоты, имеет структуру, схожую с таковой для карбена Б, мы провели встречный синтез бицикла (55) взаимодействием указанных реагентов в инертном растворителе, толуоле, с азеотропным удалением воды. Оказалось, что такая методика проведения реакции приводит к 15-кратному увеличению содержания изомера (55) в реакционной массе, но характеризуется низкой (не более 25%) конверсией исходного соединения (54). Кроме того, разделить образующуюся при этом смесь триазолопиримидинов (55) и (56) не удается ни дробной кристаллизацией, ни хроматографированием в тонком слое из-за одинаковой хроматографической подвижности этих соединений в подходящих элюирующих системах.
10 Пути модификации 4-арилиден-3-метил-
1-(6-метил-4-оксо-3,4-дигидропиримидин-2-ил)пиразол-5(4Н)-онов
Конфигурирование азинового цикла. Неудавшаяся попытка синтезировать N3-метил-аналоги арилиденпиразолонов (45 а-ж) побуждает к дальнейшему поиску путей модификации структуры последних. Одним из таковых представляется получение 4-арилиден-1-(4,6-диметилпиримидин-2-ил)-3-метилпиразол-5(4Н)-онов (57), осуществленное нами в соответствии с нижеследующей схемой:

Обработка 2-гидразино-4,6-диметилпиримидина (58) этилацетоацетатом в толуоле в условиях азеотропного удаления воды приводит к (4,6-диметилпиримидин-2-ил)гидразону этилацетоацетата (59), который, взаимодействуя с ароматическими альдегидами, дает целевые соединения (57).
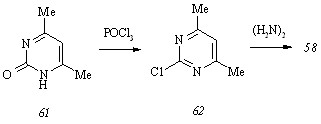
Ввиду того, что гидразинопиримидин (58) не представляется возможным синтезировать кислотным гидролизом 2-бензилиденгидразино-4,6-диметилпиримидина (60), получаемого циклоконденсацией бензилиденаминогуанидина с 2,4-пентандионом при 1300С в отсутствие растворителя, мы реализовали синтез этого субстрата с использованием традиционной схемы, включающей последовательные обменное хлорирование 4,6-диметил-2(1Н)-пиримидинона (61) и гидразинолиз 4,6-диметил-2-хлорпиримидина (62):
Нагреванием соединения (60) с разбавленной хлороводородной кислотой нами получен 2-гидразино-4-метил-6-(2-фенилэтенил)пиримидин (63), несмотря на непрерывное удаление выделяющегося бензальдегида из зоны реакции:

Низкий (около 4%) выход пиримидилпиразолона (57, Ar = Me2NC6H4) и осмоление реакционной массы при взаимодействии соединения (59) с 4-диэтиламинобензальдегидом заставляют нас обратиться к альтернативной схеме синтеза целевых соединений, основанной на использовании 1-карбоксамидино-3-метил-5(4Н)-пиразолона (64) в качестве исходного субстрата и последующей циклизации промежуточных 4-арилиден-1-карбокс-амидино-3-метил-5(4Н)-пиразолонов (65) с 2,4-пентандионом. Для осуществления означенного подхода к получению пиримидилпиразолонов (57) мы изучили взаимодействие гидрохлорида аминогуанидина (66) с этилацетоацетатом в воде в присутствии различных оснований.
Направление рассматриваемой реакции в значительной степени определяется типом используемого основания. При последовательном действии на соединение (66) гидроксидом натрия и этилацетоацетатом образуется 2,3-диамино-6-метил-4(3Н)-пиримидинон (67).
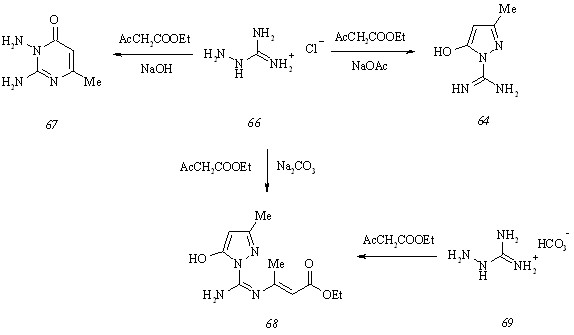
Замена гидроксида натрия менее сильным основанием, ацетатом натрия, приводит к получению целевого продукта конденсации – карбоксамидинопиразолона (64), существующего в лактимной форме с внутримолекулярной водородной связью OH···NH. Изменение направления реакции гидрохлорида аминогуанидина (66) и этилацетоацетата в присутствии ацетата натрия в сторону образования соединения (64) обусловлено протонированием выделяющейся в результате ионного обмена уксусной кислотой наиболее оснвной иминогруппы декватернизированного субстрата, что препятствует протеканию взаимодействия с участием данного реакционного центра.
Неожиданное направление конденсации гидрохлорида аминогуанидина (66) и этилацетоацетата реализуется при обработке первого карбонатом натрия и выражается в образовании функционального производного соединения (64) – этилового эфира 3-[(5-гидр-окси-3-метилпиразол-1-ил)имидоил]аминокротоновой кислоты (68). Образование этилкротоноата (68) становится объяснимым, если принять во внимание тенденцию гидрохлорида аминогуанидина (66) к разложению в присутствии карбоната натрия. Вследствие этого количество свободного аминогуанидина, подвергающегося в дальнейшем протонированию угольной кислотой с участием иминогруппы, значительно уменьшается, и в реакционной массе возникает избыток этилацетоацетата. Растворяясь в воде, диоксид углерода частично превращается в угольную кислоту, которая связывается с аминогуанидином и дает карбонат аминогуанидина (69). Соединение (69), последовательно взаимодействуя с избытком этилацетоацетата, приводит к этилкротоноату (68). Подтверждением этой гипотезы является конденсация карбоната аминогуанидина (69) и этилацетоацетата в мольном соотношении 1:2, протекающая с образованием продукта (68).
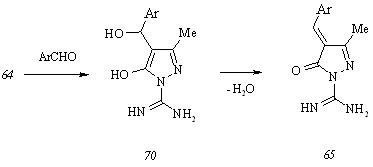
Конденсация карбоксамидинопиразолона (64) с 4-диметиламинобензальдегидом в кипящем метаноле приводит к образованию двух соединений, одно из которых характеризуется значением хроматографического индекса, равным таковому для карбоксамидинопиразолона (64), но обнаруживается в видимом свете при элюировании пластины. Попытка разделения смеси веществ, выделенной после испарения растворителя, кристаллизацией из подобранного растворителя не увенчалась успехом и привела лишь к уменьшению величин их хроматографических индексов. Мы предположили, что компонентом выделенной смеси является не исходный карбоксамидинопиразолон (64), проявляющийся на пластине исключительно при внесении ее в зону УФ облучения, но промежуточный 5-гидрокси-4-[-гидрокси--(4-диметиламинофенил)метил]-1-карбоксамидино-3-метилпир-азол (70, Ar = Me2NC6H4), не подвергшийся исчерпывающей дегидратации до ожидаемого 4-(4-диметил-аминобензилиден)-1-карбоксамидино-3-метилпиразол-5(4Н)-она (65, Ar = Me2NC6H4):
В случае корректности выдвинутого нами предположения для образования соединения (65, Ar = Me2NC6H4) необходимо соблюдение условий, обеспечивающих непрерывное удаление воды из зоны реакции. Поскольку карбоксамидинопиразолон (64) обладает низкой растворимостью в средах, способных связывать выделяющуюся воду в устойчивый азеотроп, мы прибегли к осуществлению конденсации между соединением (64) и 4-диметиламинобензальдегидом при температурах 150-1600С. Для исключения присутствия непрореагировавшего карбоксамидинопиразолона (64) среди продуктов реакции, полученный плав суспендировали в водном растворе аммиака, однако выделенный таким образом окрашенный продукт оказался весьма неустойчивым соединением и подвергался разложению при кристаллизации из подходящих растворителей с образованием маслянистой полимерной субстанции.
Взаимодействие карбоксамидинопиразолона (64) с 2,4-пентандионом также сопровождается трудностями осуществления. Нагреванием смеси исходных реагентов в присутствии дегидратирующего агента, пентаоксида фосфора, нам не удалось получить ни 5-гидрокси-3-метил-1-[N-(4-оксопентан-2-ен-2-ил)карбоксамидино]пиразол (71), ни 5-гидр-окси-1-(4,6-диметилпиримидин-2-ил)-3-метилпиразол (72). Небольшие количества первого образуются лишь при кипячении карбоксамидинопиразолона (64) в избытке дикетона:

Жесткие условия синтеза и низкий выход оксопентена (71) указывают на высокую прочность внутримолекулярной связи в исходном субстрате (64). Чтобы предотвратить влияние хелатообразования на реакционную способность амидиновой группы, мы провели взаимодействие натриевой соли карбоксамидинопиразолона (64) с 2,4-пентандионом в абсолютном этаноле, который позволяет локализовать анионный центр, возникающий на атоме кислорода гетероцикла, посредством образования водородных связей и тем самым исключить иминогруппу из участия в потенциальном процессе переноса протона. Несмотря на обоснованный выбор растворителя, подобный вариант осуществления гетероциклизации не привел к ожидаемому результату. Из реакционной массы, предварительно не подвергнутой нейтрализации, нами выделено соединение (64). Это обстоятельство убедительно свидетельствует о депротонировании молекулы дикетона анионом карбоксамидинопиразолона (64), выполняющим функцию основания:
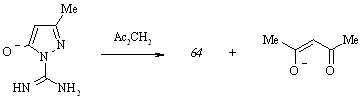
Конфигурирование фрагмента -арил-,-ненасыщенного кетона. Другим маршрутом модификации структуры арилиденпиразолонов (45 а-ж) представляется трансформация фрагмента -арил-,-ненасыщенного кетона их молекулы. Для реализации этого маршрута мы синтезировали 4-ариламинометилен-3-метил-1-(6-метил-4-оксо-3,4-дигидропири-мидин-2-ил)пиразол-5(4Н)-оны (73 а-к) трехкомпонентной конденсацией пиразолона (47), триэтоксиметана и ароматического амина в отсутствие растворителя при 800С по схеме:

В противоположность описанному способу получения соединения (47), основанному на циклизации промежуточного пиримидилгидразона (46) под действием основания, нами предложен таковой, который позволяет генерировать субстрат (47) непосредственным взаимодействием гидразинопиримидинона (44) с этилацетоацетатом в воде в отсутствие катализатора деэтерификации.
Введение вторичной аминогруппы во фрагмент -арил-,-ненасыщенного кетона арилиденпиразолонов (45 а-ж) приводит к значительному увеличению противотуберкулезной активности возникающих таким образом ариламинометиленпиразолонов (73 а-к) (табл. 5).
Таблица 5 – Количественная оценка биологической активности
4-ариламинометилен-3-метил-1-(6-метил-4-оксо-3,4-дигидропиримидин-2-ил)-
пиразол-5(4Н)-онов (73 а-к)
| № соед. | Ar | Минимальная концентрация, обеспечивающая ингибирование биообъекта на 100% IC100, мкг/мл | |
| Esherichia coli | Mycobacterium tuberculosis | ||
| 73 а | Ph | - | 12.5 |
| 73 б | 4-MeC6H4 | - | 12.5 |
| 73 в | 4-EtC6H4 | - | - |
| 73 г | 4-FC6H4 | 12.5 | 12.5 |
| 73 д | 4-ClC6H4 | 12.5 | 12.5 |
| 73 е | 4-BrC6H4 | - | 50 |
| 73 ж | 4-IC6H4 | - | 12.5 |
| 73 з | 3-Me-4-BrC6H3 | - | 12.5 |
| 73 и | 3,4-Cl2C6H3 | - | |
| 73 к | 3-Cl-4-MeOC6H3 | - | |
Большинство соединений (73 а-к) ингибируют рост клеток культуры Mycobacterium tuberculosis штамма H37Rv в концентрациях, значительно меньших по сравнению с определенными для арилиденпиразолонов (45 а-ж). Вместе с тем, приведенные значения IC100 свидетельствуют о малой чувствительность данного штамма к природе заместителя в пара-положении кольца. Кроме того, заметная склонность к подавлению роста клеток бактериального происхождения Esherichia coli наблюдается в случае отдельных представителей ариламинометиленпиразолонов (73 а-к).
11 Реакционная способность 1-(пиримидин-2-ил)пиразол-5(4Н)-онов
В то время, как объектом атаки ароматическими альдегидами служит положение 4 пятичленного кольца пиразолона (47), направление взаимодействия этого субстрата с винилогами карбонильных соединений в условиях реакции Михаэля обусловливается последовательностью процессов ионизации -метиленкетонных фрагментов гетероциклов, входящих в его состав. Для предсказания структуры аддукта, полученного присоединением 5-бензилиден-2,4,6(1Н,3Н,5Н)-пиримидинтриона (74) к пиразолону (47), мы изучили строение моноионизированной формы субстрата.
Копланарное расположение атомов и близость расстояний между ними способствуют возникновению внутримолекулярных связей типов C=O···HN и NH···N, которые, в свою очередь, вызывают образование равновесной смеси таутомеров (47 а) и (47 б) в чистом этаноле. Прибавление к раствору этилата натрия вызывает сдвиг равновесия в сторону таутомера (47 а) и приводит к разрушению водородной связи NH···O и последующему образованию ионизированной формы А:
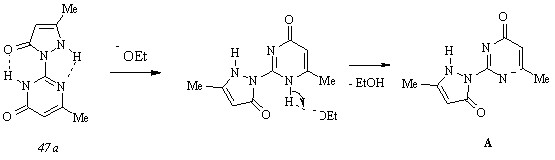
По мере увеличения мольного содержания основания происходит протонирование ионизированной формы А путем миграции протона NH группы пиразольного цикла к пиримидиновому и заключительное образование таутомера (47 б):
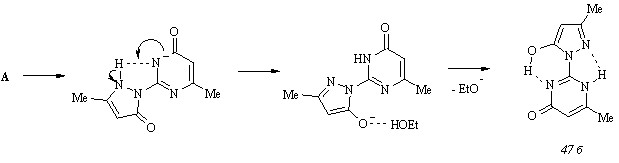
Варьирование температуры не оказывает заметного влияния на процесс ионизации пиразолона (47), и этот факт позволяет утверждать о неизменности структуры ионизированной формы А в широком диапазоне температур, в том числе при температуре исследуемого взаимодействия. Ионизация пиримидинового цикла влечет за собой повышение его нуклеофильности, определяющее преимущественное присоединение бензилиденбарбитуровой кислоты (74) к СН-кислотному центру диазиновой составляющей молекулы пиразолона (47). Действительно, при конденсации исходных соединений (47) и (74) в этаноле в присутствии этилата натрия при 800С нами выделен единственный продукт реакции – (2,4,6-триоксо-1,2,3,4,5,6-гексагидропиримидин-5-ил)[2-(5-гидрокси-3-метилпир-азол-1-ил)-6-метил-4-оксо-3,4-дигидропиримидин-5-ил]фенилметан (75):
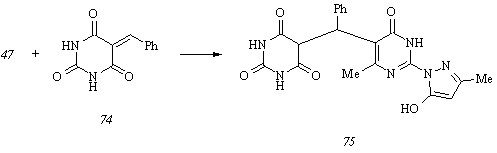
Исследование его гидролитической устойчивости показывает выраженную тенденцию к разложению уже при суспендировании в воде: продуктами гидролиза соединения (75) являются пиразолон (47), барбитуровая кислота и бензальдегид.
12 Производные 1-(пиримидин-4-ил)пиразол-5(4Н)-она в реакции Кневенагеля
Синтез арилиденпиразолонов (45 а-ж) способом а или традиционным путем из пиразолона (47) и ароматических альдегидов с приемлемыми (30-70%) выходами и неприменимость означенных подходов к получению пиримидилпиразолонов (57) позволяют выдвинуть предположение о затрудненной дегидратации промежуточного 5-гидрокси-4-[-гидрокси--(4-диметил-амино-фенил)-метил]-1-(4,6-диметилпиримидин-2-ил)-3-метил-пиразола, предшественника пиримидилпиразолона (57, Ar = Me2NC6H4), вследствие образования внутримолекулярной водородной связи между копланарно расположенными гидроксигруппой пиразольного цикла и одним из атомов азота ядра пиримидина:

Чтобы исключить или, по крайней мере, минимизировать негативное влияние вышеуказанного способа взаимной ориентации на процесс образования арилиденпиразолонов типа (45 а-ж), мы исследовали возможность получения 4-арилиден-3-метил-1-(6-метил-2-метилтиопиримидин-4-ил)пиразол-5(4Н)-онов (76) в условиях реакции Кневенагеля. Необходимый для этого 5-гидрокси-3-метил-1-(6-метил-2-метилтиопиримидин-4-ил)пиразол (77) синтезирован в соответствии со схемой:

Взаимодействие 4-гидразино-6-метил-2-метилиопиримидина (78) с этилацетоацетатом в 1-бутаноле в условиях азеотропного удаления воды дает (6-метил-2-метилтио-пиримидин-4-ил)гидразон этилацетоацетата (79), который при обработке гидроксидом калия в этаноле превращается в гидроксипиразол (77). Получение гидразина (78) из соответствующего 6-метил-2-метилтио-4-хлорпиримидина (80) потребовало от нас разработки оригинального способа обменного хлорирования метилтиоэфира (1) пентахлоридом фосфора при 75-1350С, так как предложенные методы не позволяли выделять субстрат (80) удовлетворительной степени чистоты и с препаративным выходом.
Как и пиримидилгидразон (46), соединение (77), взаимодействуя с ароматическими альдегидами в этаноле при 40-500С в присутствии диэтиламина, дает два типа продуктов, строение которых определяется электронным эффектом заместителя в карбонильной компоненте реакции. При конденсации гидроксипиразола (77) с 4-диметиламинобензальде-гидом (R1 = 4-Me2NC6H4) мы получили 4-(4-диметиламинобензилиден)-3-метил-1-(6-ме-тил-2-метилтиопиримидин-4-ил)пиразол-5(4Н)-он (76 а, Ar = 4-Me2NC6H4). Замена использованного альдегида 4-нитробензальдегидом (R2 = 4-O2NC6H4) и проведение реакции в тех же условиях приводит к образованию диэтиламмониевой соли бис[5-гидрокси-3-метил-1-(6-метил-2-метилтиопиримидин-4-ил)пиразол-4-ил](4-нитрофенил)метана (81) даже с учетом использования эквимольного соотношения компонентов.
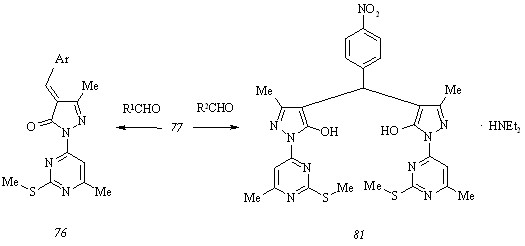
Нейтрализацией диэтиламмониевой соли (81) уксусной кислотой может выделен арилбиспиразолилметан в виде свободного основания.
Электронодонорные свойства пара-диалкиламинозамещенного фенильного кольца проявляются в некоторых случаях настолько сильно, что препятствуют дегидратации промежуточно образующихся 4-(-гидрокси-4-диалкиламинобензил)-3-метил-1-(6-метил-2-метилтиопиримидин-4-ил)пиразол-5(4Н)-онов (82). В сравнении со смесью соединения (76 а) и интермедиата (82, Alk = Me), разделяемой однократной кристаллизацией, смесь, образующуюся при взаимодействии гидроксипиразола (77) с 4-диэтиламинобензальде-гидом и состоящую из -гидроксибензилпиразолона (82, Alk = Et) и 4-(4-диэтиламино-бензилиден)-3-метил-1-(6-метил-2-метилтиопиримидин-4-ил)пиразол-5(4Н)-она (76 б, Ar = 4-Et2NC6H4), который обнаруживается в виде ярко окрашенного пятна уже при элюировании хроматографической пластины, не удается разделить на компоненты, применяя указанную процедуру. Более того, неэффективными являются как выдерживание смеси продуктов (82, Alk = Et) и (76 б) при 1000С над дегидратирующим агентом, пентаоксидом фосфора, в вакууме, так и нагревание в бензоле с водоотделителем в присутствии каталитического количества концентрированной серной кислоты. Последний прием, хотя и позволяет подтвердить наше предположение относительно нахождения в исходной смеси -гидроксибензилпиразолона (82, Alk = Et) фактом выделения воды, не приводит к полной трансформации этого соединения в арилиденпроизводное (76 б).
13 Электрофильное замещение
в 5-гидрокси-3-метил-1-(6-метил-2-метилтиопиримидин-4-ил)пиразоле
Образование соединения (76 а) при конденсации гидроксипиразола (77) с 4-диметил-аминобензальдегидом позволяет рассматривать данный субстрат как активированный -метиленкетон и в этой связи надеяться на не менее успешное его взаимодействие с другими электрофильными реагентами или с таковыми, способными генерировать электрофильную частицу в зоне реакции. Для того, чтобы убедиться в этом, мы исследовали поведение соединения (77) по отношению к некоторым галогенам, арилдиазонийхлоридам, а также при взаимодействии с аминометиленирующим, гидроксиметилирующим и нитрозирующим агентами.
Общей особенностью протекания рассматриваемых ниже реакций электрофильного замещения в гидроксипиразоле (77) служит исключительное участие в них атома С4 пиразольного цикла. Этому способствует специфическая делокализация -электронной плотности, приводящая к компенсации частичного положительного заряда на указанном атоме и повышению таким образом его нуклеофильности:

Отсутствие активирующих заместителей типа оксигрупп препятствует увеличению нуклеофильности и вследствие этого затрудняет атаку электрофилами псевдоароматического положения пиримидинового кольца соединения (77).
Трансформации гидроксипиразола (77) (для компактности изображения введем обозначение R = 6-метил-2-метилтиопиримидин-4-ил) под действием использованных электрофильных реагентов можно разделить на две группы. Первую составляют реакции, приводящие к образованию продуктов монозамещения – галогенирование, азосочетание и аминометиленирование, ко второй относятся взаимодействия, позволяющие получать продукты мостиковой структуры – гидроксиметилирование и нитрозирование.
Прямое галогенирование. Бромирование соединения (77) бромом в тетрахлорметане при 250С дает смесь 4-бром-5-гидрокси-3-метил-1-(6-метил-2-метилтиопиримидин-4-ил)пир-азола
[1] (83) и неидентифицируемого соединения, из которой целевой бромпиразол (83) выделяется в хроматографически чистом виде с низким выходом после дробной кристаллизации.
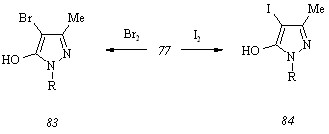
При йодировании гидроксипиразола (77) йодом в 10%-ном водном растворе гидроксида натрия образования других продуктов, кроме 5-гидрокси-4-йод-3-метил-1-(6-метил-2-метилтиопиримидин-4-ил)пиразола (84) не происходит, но имеет место значительное осмоление реакционной массы, что приводит к выделению целевого соединения в хроматографически чистом виде с выходом не более 15%.
Азосочетание. При взаимодействии гидроксипиразола (77) с бензолдиазонийхлоридом в водном растворе гидроксида натрия при температуре не выше 100С образуется 3-метил-1-(6-метил-2-метилтиопиримидин-4-ил)-4-фенилгидразонопиразол-5(4Н)-он (85), обладающий внутримолекулярной водородной связью:

Аминометиленирование. Нагревание гидроксипиразола (77) со смесью триэтоксиметана и фениламина в этаноле при 500С протекает аномальным путем, приводя к образованию неожиданного продукта – 4-гидроксиметилен-3-метил-1-(6-метил-2-метилтиопиримидин-4-ил)пиразол-5(4Н)-она (86) после обработки реакционной массы водным раствором аммиака:

Исключение растворителя из взаимодействия соединения (77) с аминометиленирующим агентом и проведение реакции при 800С позволяет получить целевой продукт – 5-гидрокси-3-метил-1-(6-метил-2-метилтио-пиримидин-4-ил)-4-фенилиминометиленпир-азол (87).
Гидроксиметилирование. При конденсации соединения (77) с формальдегидом, взятым в эквимольном количестве по отношению к субстрату, в водном растворе гидроксида натрия при 250С происходит образование бис[5-гидрокси-3-метил-1-(6-метил-2-метилтио-пиримидин-4-ил)пиразол-4-ил]метана (88):

Нитрозирование. Обработка гидроксипиразола (77) водным раствором нитрита натрия при 250С в разбавленной уксусной кислоте приводит к образованию 3-метил-1-(6-метил-2-метилтиопиримидин-4-ил)-4-[5-гидрокси-3-метил-1-(6-метил-2-метилтиопиримидин-4-ил)пиразол-4-ил]иминопиразол-5(4Н)-она (89):
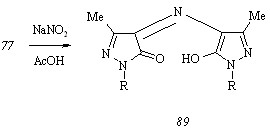
Выделение продуктов мостиковой структуры, например, биспиразолилметана (88), обусловлено делокализацией частичного положительного заряда, возникающего на -углеродном атоме промежуточного цвиттериона А, путем присоединения второй молекулы ионизированного гидроксипиразола (77), выполняющего функцию нуклеофила. Косвенным подтверждением корректности предложенной схемы служит выход соединения (88), низкая величина (менее 20%) которого указывает на стабилизацию цвиттериона А не только указанным способом, но и обратимым присоединением гидроксил-иона с образованием гибрида анионов Б и В, могущего проявлять некоторую устойчивость вследствие гидратированности молекулами реакционной среды:
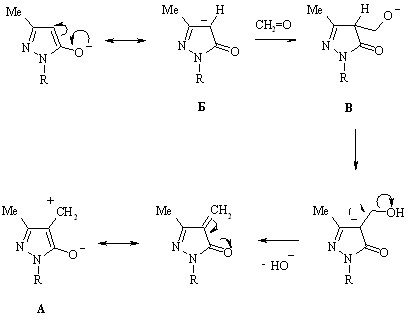
14 Влияние свойств среды на положение и подвижность лабильного протона
в 5-гидрокси-3-метил-1-(6-метил-2-метилтиопиримидин-4-ил)пиразоле
При изучении реакционной способности гидроксипиразола (77) по отношению к электрофильным реагентам мы без доказательств приписали субстрату лактимную форму существования, не учитывая возможности ее трансформации в лактамную под влиянием специфической сольватации. Как явствует из нижеизложенного, подобная позиция оправдана лишь в тех случаях, когда речь ведется о растворах соединения (77) в спиртах. С целью подтвердить или опровергнуть правомерность отнесения гидроксипиразола (77) к гетероциклическим енолам независимо от сольватных свойств среды или величины ее диэлектрической проницаемости r нами исследовано влияние этих факторов на положение и подвижность лабильного протона соединения (77) в растворителях различной природы методом УФ спектроскопии.
Поглощение УФ света гидроксипиразолом (77) в этаноле выражается кривой с двумя максимумами 248 и 310 нм, из которых только длинноволновый претерпевает гипсохромное смещение при прибавлении к раствору близкого к эквимольному количества основания, этилата натрия. Изосбестическая точка, обнаруживаемая в спектрах растворов с отличным содержанием основания, свидетельствует о частичной ионизации соединения (77) даже в чистом этаноле:
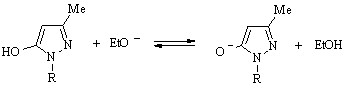
Функцию основания в растворе гидроксипиразола (77) с нулевым содержанием этилата натрия выполняет растворитель, вызывая эффект ионизации посредством образования водородной связи с гидроксигруппой и последующей миграции входящего в ее состав протона к молекуле среды:

Замена этанола водой, склонной к образованию водородных связей в значительно большей степени, приводит к неожиданному на первый взгляд сдвигу длинноволнового максимума полосы поглощения к 305 нм. Вместе с тем наблюдаемое изменение в спектре соединения (77) хорошо согласуется с выдвинутым утверждением о его ионизации, инициирумой растворителем, с образованием мезомерного комплекса А, в котором максимальному разделению зарядов способствуют электронодонорные свойства алкильного радикала. На менее выгодное возникновение альтернативного комплекса Б между молекулами гидроксипиразола (77) и воды, характеризующегося отсутствием возможности подобной компенсации дефицита электронной плотности на атоме кислорода и обладающего вследствие этого повышенной потенциальной энергией, указывает не только гипсохромное смещение максимума, но и существенное [ ~ 2680 л/(моль·см)] уменьшение интенсивности рассматриваемой полосы поглощения соединения (77):
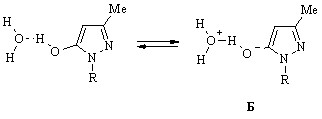
Содержание комплекса Б в растворе едва ли превышает 1%, поскольку той же величиной, полученной исходя из значения pKa гидроксипиразола (77), описывается степень ионизации этого соединения при рН 7, вследствие чего равновесие между гидратированными электронейтральной и ионизированной формами гидроксипиразола (77) в нейтральном растворе практически нацело сдвинуто в сторону первой.
Участие электронодонорного алкильного радикала в компенсации положительного заряда в мезомерном комплексе А иллюстрируется возрастанием молярного коэффициента поглощения длинноволновой полосы в спектрах соединения (77) в высших спиртах, происходящего на фоне сохранения ее положения в области 310±2 нм.
Применение вместо спиртов, типичных амфипротонных растворителей, высокополярного ацетонитрила, являющегося акцептором водородной связи, вносит заметные изменения в спектр гидроксипиразола (77), которые выражаются в батохромном сдвиге коротковолновой полосы поглощения к 260 нм, смещении полосы с максимумом 310 нм в сторону меньших длин волн и появлении плеча вблизи 340 нм. Принимая во внимание оснвные свойства растворителя, наличие перегиба кривой поглощения можно объяснить n * переходом в ионизированной лактамной форме соединения (77), образующейся в результате перераспределения электронной плотности в мезомерном комплексе В, а таутомеризацию лактима, существующего в менее полярном (r 24.55) этаноле, в лактам – стабилизацией последнего как формы повышенной полярности в более полярном (r 35.94) ацетонитриле:
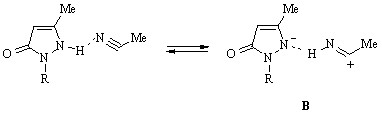
Прибавление триэтиламина к раствору гидроксипиразола (77) в этом растворителе вызывает обратный сдвиг только коротковолновой полосы в область 245-250 нм. Несмотря на отсутствие изосбестической точки в спектрах соединения (77) и триэтиламина варьируемых соотношений, смещение максимума обсуждаемой полосы и близость ее положения и интенсивности аналогичным параметрам полосы, отвечающей поглощению смеси лактимной формы и ее аниона в этаноле в присутствии этилата натрия, свидетельствуют о переносе заряда с атома азота на атом кислорода и установлении равновесия между ионизированными формами гидроксипиразола (77), сольватированными биполярным ацетонитрилом:
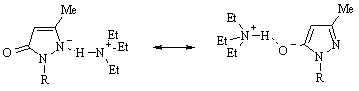
Переход от ацетонитрила к апротонному тетрахлорметану сопровождается сохранением положения длинноволновой полосы и некоторым увеличением ее интенсивности. Наряду с симметричностью обсуждаемой полосы присущий ей гиперхромный эффект указывает, во-первых, на неподверженность соединения (77) таутомерной перегруппировке в лактам и, во-вторых, на отсутствие у этого соединения склонности к образованию меж- и внутримолекулярных водородных связей N···HO. Причиной подобного поведения гидроксипиразола (77) служат, с одной стороны, неспособность малополярного (r 2.2) тетрахлорметана к стабилизации полярных ассоциатов с водородной связью, а с другой – недостаточной основностью атомов азота пиразольного и пиримидинового колец. Образование межмолекулярного ассоциата происходит только при использовании оснований с высоким значением рКа сопряженной кислоты типа триэтиламина и не сопровождается возникновением равновесия с мезомерным комплексом Г, характеризующимся значительной степенью поляризации связей и разделения зарядов:
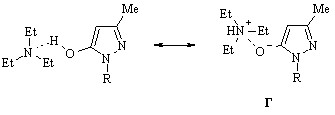
На это указывают подобие кривых поглощения индивидуального соединения (77) и его смеси с эквимольным количеством триэтиламина в гексане, а также несущественное (не более 4-5%) увеличение молярных коэффициентов поглощения обеих полос в последнем случае.
15 Синтез 1-(6-метил-4-оксо-3,4-дигидропиримидин-2-ил)тиосемикарбазидов
Помимо циклизации в производные 2-(пиразол-1-ил)пиримидина, гидразинопиримидины (44) и (54) в виде гидрохлоридов способны претерпевать трансформацию в 1-(6-метил-4-оксо-3,4-дигидропиримидин-2-ил)тиосемикарбазиды (90 а, б) под действием роданида калия:
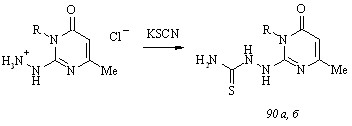
R = H (a), Me (б)
Реакция протекает в воде при температуре 250С. Ввиду сложности обнаружения тиокарбамидной группы спектральными методами мы судили о ее присутствии в молекулах тиосемикарбазидов (90 а, б) с помощью качественной реакции, заключавшейся в выделении диоксида серы из их водных растворов, подвергнутых обработке раствором перманганата калия.
Наличие тиокарбамидной группы позволяет рассчитывать на возможность использования соединений (90 а, б) в синтезе 1-(пиримидин-2-ил)-2-(тиазол-2-ил)гидразинов методом Ганча.
16 Конденсация 1-(6-метил-4-оксо-3,4-дигидропиримидин-2-ил)тиосемикарбазидов
с арилхлорметилкетонами
Взаимодействие тиосемикарбазидов (90 а, б) с арилхлорметилкетонами протекает неоднозначно. Кипячение соединения (90 а) с фенацилхлоридом в 1-бутаноле с азеотропным удалением воды (способ а) приводит к смеси ожидаемого продукта реакции Ганча, гидрохлорида 1-(6-метил-4-оксо-3,4-дигидропиримидин-2-ил)-2-(4-фенилтиазол-2-ил)-гидразина (91) и смолообразных веществ, из которой выделить соединение (91) в хроматографически чистом виде не представляется возможным. При нагревании компонентов до 1000С в отсутствие растворителя (способ б) имеет место образование нового соединения, разлагающегося в процессе кристаллизации из подобранных растворителей.
В отличие от тиосемикарбазида (90 а) его N-метилированный аналог (90 б) реагирует с фенилхлорметилкетоном способом а, давая 1-(3,6-диметил-4-оксо-3,4-дигидропирими-дин-2-ил)-2-(4-фенилтиазол-2-ил)гидразин (92 a) в виде гидрохлорида спектральной степени чистоты:
Ar = Ph (a), 4-ClC6H4 (б), 3,4-(HO)2C6H3 (в)
Аналогично фенацилхлориду в конденсацию с тиосемикарбазидом (90 б) вступают другие арилхлорметилкетоны: с 4-хлорфенацилхлоридом субстрат образует гидрохлорид 1-(3,6-диметил-4-оксо-3,4-дигидропиримидин-2-ил)-2-[4-(4-хлорфенил)тиазол-2-ил]гидр-азина (92 б) при осуществлении реакции способом б, с 3,4-дигидроксифенацилхлоридом – 2-[4-(3,4-дигидроксифенил)тиазол-2-ил]-1-(3,6-диметил-4-оксо-3,4-дигидропиримидин-2-ил)гидразин (92 в) в виде гидрохлорида способом а. Особенностью выделения последнего соединения после очистки кристаллизацией из ДМФА служит возникновение сольвата состава 1:1 следующей структуры:
Выделение 1-пиримидил-2-тиазолилгидразинов (92 а-в) в виде гидрохлоридов требует оценки вклада каждого из гетероциклов в процесс кватернизации. Для разрешения этого вопроса мы прибегли к сопоставлению УФ спектра соединения (92 а) со спектрами модельных соединений, гидрохлоридов 2-амино-3,6-диметил-4(3Н)-пиримидинона (93) и 2-амино-4-фенилтиазола (94), в предположении, что электронные переходы типа n * в гидрохлоридах 1-пиримидил-2-тиазолилгидразинов (92 а-в) ограничиваются пределами системы связей HNex-C2 =N1 пиримидинового и тиазольного колец. Выявленная картина поглощения УФ света соединениями (92 a), (93) и (94) свидетельствует об ошибочности нашего предположения относительно локализации протона у одного из циклических атомов азота 1-пиримидил-2-тиазолилгидразина (92 а): его спектр представляет собой кривую с двумя перегибами, из которых только один, располагающийся вблизи 260 нм, совпадает с плечом в спектре гидрохлорида аминотиазола (94). Помимо ароматических систем в указанной области наблюдается поглощение протонированных 2-амино-4(3Н)-пиримидинонов, характеризующихся имино-лактамной формой существования. С учетом высказанного, а также принимая во внимание то обсоятельство, что возникновение цепи сопряжения в молекуле 1-пиримидил-2-тиазолилгидразина (92 а) возможно лишь в случае взаимозависимых таутомерных перегруппировок его гетероциклических фрагментов, можно считать, что строение рассматриваемого соединения выражается гибридом резонансных структур А и Г:

Подтверждением подверженности 1-пиримидил-2-тиазолилгидразина (92 а) описанному типу таутомеризации служит наличие в его спектре длинноволнового плеча низкой интенсивности около 310 нм, отвечающего n * электронным переходам в цепи сопряжения, включающей промежуточные протонированные иминоформы Б и В.
ВЫВОДЫ
1. Результатом разработки унифицированных путей синтеза и изучения химических трансформаций и таутомерных превращений 2-амино- и 2-гидразино-4(3Н)-пиримиди-нонов послужило получение ряда моно- и полициклических производных 2- и 4-амино(гидразино)пиримидинов, проявляющих выраженное биологическое действие in vitro.
2. Предложен унифицированый способ бромирования in situ трудно доступных в кристаллическом состоянии 2-алкил(циклоалкил-, аралкил)амино-6-метил-4(3Н)-пирими-динонов.
3. Установлен механизм таутомеризации 2-амино-6-метил-4(3Н)-пиримидинонов, обусловленной специфической гидратацией и высокой полярностью среды, через промежуточное образование ассоциата цвиттерионной структуры.
4. Обнаружена внутримолекулярная перегруппировка продукта О-арилкарбамои-лирования 2-(2-гидроксиэтил)амино-6-метил-4(3Н)-пиримидинона в условиях кислотного катализа.
5. Выявлено направление циклоконденсации (4-оксо-3,4-дигидропиримидин-2-ил)-гидразонов этилацетоацетата и ароматических альдегидов в зависимости от электронных эффектов заместителей в ароматическом кольце.
6. Установлен механизм аномальной циклизации (4-оксо-3,4-дигидропиримидин-2-ил)гидразонов этилацетоацетата с фиксированной конфигурацией гетероядра в 1,2,4-триазоло[1,5-a]пиримидин-5(4Н)-оны путем термической деструкции алифатического фрагмента.
7. Обнаружена внутримолекулярная миграция илиденового остатка 2-бензилиден-гидразино-4,6-диметилпиримидина, катализируемая минеральной кислотой, к метильной группе при гидролитическом расщеплении субстрата.
8. Изучены приоритетные направления электрофильного замещения в 1-пиримидил-пиразол-5(4Н)-онах, определяемые электронными эффектами заместителей в азиновом кольце.
9. Предложена молекулярная структура гидрохлоридов 1-(пиримидин-2-ил)-2-(тиазол-2-ил)гидразинов, стабилизирующаяся за счет взаимозависимых таутомерных перегруппировок гетероциклических фрагментов их молекул.
10. Посредством расширенного биологического скрининга моно- и полициклических производных 2- и 4-амино(гидразино)пиримидинов выявлены две группы сильных ингибиторов роста клеточных культур бактериального и микобактериального происхождений, структуры которых принципиально отличаются от таковых для известных лекарственных препаратов аналогичного действия.
Основное содержание работы изложено в следующих публикациях:
1. Еркин А.В., Крутиков В.И., Павлович Н.И. Унифицированный подход к синтезу 2-замещенных 6-метилизоцитозинов и их 5-бромпроизводных // Журнал общей химии. 2003. Т. 73. Вып. 3. С. 494-498.
2. Еркин А.В., Крутиков В.И., Чубраев М.А. Синтез производных 2-(пиразол-1-ил)пиримидина циклоконденсацией (6-метил-4-оксо-3,4-дигидропиримидин-2-ил)гидр-азона этилацетоацетата с ароматическими альдегидами // Журнал общей химии. 2004. Т. 74. Вып. 3. С. 466-471.
3. Еркин А.В., Крутиков В.И. Модифицированный синтез и биологическая активность производных 2-(пиразол-1-ил)пиримидинов // Конференция «Наукоемкие химические технологии-2004»: Тез. докл. – Волгоград, Изд-во МИХТ, 2004. С. 245-247.
4. Еркин А.В., Крутиков В.И. Полифункциональные производные изоцитозина. Влияние гидратации на прототропную таутомерию 2-(2-гидроксиэтил)амино-6-метил-4-оксо-пиримидина // Журнал общей химии. 2005. Т. 75. Вып. 4. С. 677-682.
5. Косых А.В., Еркин А.В., Крутиков В.И. Структура и кислотный гидролиз карбамоильных производных полифункциональных изоцитозинов // VIII Молодежная научная школа-конференция по органической химии: Тез. докл. – Казань, Изд-во центра инновац. технол., 2005. С. 216.
6. Еркин А.В., Крутиков В.И., Косых А.В. Полифункциональные производные изоцитозина. I. Внутримолекулярная перегруппировка 6-метил-4-оксо-2-[2-(фенилкарбамоил-окси)этил]аминопиримидина // Журнал общей химии. 2005. Т. 75. Вып. 11. С. 1898-1901.
7. Еркин А.В., Крутиков В.И. Полифункциональные производные изоцитозина. II. Региоселективность метилирования 2-(2-гидроксиэтил)амино-6-метил-4-оксопиримидина в различных средах // Журнал общей химии. 2006. Т. 76. Вып. 3. С. 501-505.
8. Еркин А.В., Крутиков В.И., Андреева Е.В. Аномальная циклизация (3,6-метил-4-оксо-3,4-дигидропиримидин-2-ил)гидразона этилацетоацетата // XI Международная конференция «Наукоемкие химические технологии-2006»: Тез. докл. – Самара, 2006. С. 134-135.
9. Еркин А.В., Крутиков В.И. Аномальная циклизация (3,6-метил-4-оксо-3,4-дигидро-пиримидин-2-ил)гидразона этилацетоацетата // Журнал общей химии. 2007. Т. 77. Вып. 1. С. 133-136.
10. Еркин А.В., Крутиков В.И. 4-Ариламино-2-(2-ацетоксиэтил)амино-6-метилпири-мидины: синтез, деацетилирование и биологическая активность // Журнал общей химии. 2007. Т. 77. Вып. 11. С. 1887-1892.
11. Еркин А.В., Крутиков В.И. Модифицированный синтез некоторых 1-(пиримидин-2-ил)-3-метил-4-арилиденпиразол-5(4Н)-онов // Журнал общей химии. 2008. Т. 78. Вып. 2. С. 324-327.
12. Еркин А.В., Крутиков В.И., Смирнова Е.Б. Синтез и антимикобактериальная активность 2-замещенных 4-ариламино-6-метилпиримидинов // Журнал общей химии. 2008. Т. 78. Вып. 10. С. 1708-1712.
13. Еркин А.В., Крутиков В.И. Особенности кислотного гидролиза 2-бензилиден-гидразино-4,6-диметилпиримидина // Журнал общей химии. 2009. Т. 79. Вып. 4. С. 699-700 (письмо в Редакцию).
14. Еркин А.В., Крутиков В.И. Образование, строение и гетероциклизация продуктов конденсации аминогуанидина с этилацетоацетатом // Журнал общей химии. 2009. Т. 79. Вып. 6. С. 1032-1037.
15. Еркин А.В., Крутиков В.И. 6-Метил-2-(3-метил-5-оксо-2,5-дигидропиразол-1-ил)-пиримидин-4(1Н)-он как СН-кислота в реакции Михаэля // Журнал общей химии. 2009. Т. 79. Вып. 7. С. 1168-1174.
16. Еркин А.В., Крутиков В.И. Влияние локализации атома галогена на уровень антимикобактериальной активности 2-амино-4-ариламино-6-метилпиримидинов // Журнал общей химии. 2010. Т. 80. Вып. 4. С. 657-663.
17. Еркин А.В., Крутиков В.И. Потенциальные антимикобактериальные препараты на основе производных изоцитозина // Научно-практическая конференция, посвященная 182-й годовщине образования Санкт-Петербургского государственного технологического института (технического университета): Тез. докл. – СПб, Изд-во СПбГТИ(ТУ), 2010. С. 55.
18. Еркин А.В., Крутиков В.И. Производные 1-(пиримидин-4-ил)пиразол-5(4Н)-онов. I. Синтез 5-гидрокси-3-метил-1-(6-метил-2-метилтиопиримидин-4-ил)пиразола и особенности его участия в реакции Кневенагеля // Журнал общей химии. 2011. Т. 81. Вып. 2. С. 294-298.
19. Еркин А.В., Крутиков В.И. Синтез и молекулярная структура 1-(пиримидин-2-ил)-2-(4-арилтиазол-2-ил)гидразинов // Журнал общей химии. 2011. Т. 81. Вып. 8. С. 1354-1359.
20. Еркин А.В., Крутиков В.И. Производные 1-(пиримидин-4-ил)пиразол-5(4Н)-онов. II. Электрофильное замещение в 5-гидрокси-3-метил-1-(6-метил-2-метилсульфанил-пиримидин-4-ил)пиразоле // Журнал общей химии. 2011. Т. 81. Вып. 8. С. 1360-1366.
21. Еркин А.В., Крутиков В.И. Производные 1-(пиримидин-4-ил)пиразол-5(4Н)-онов. III. Влияние свойств среды на локализацию и подвижность лабильного протона в 5-гидрокси-3-метил-1-(6-метил-2-метилтиопиримидин-4-ил)пиразоле // Журнал общей химии. 2011. Т. 81. Вып. 9. С. 1552-1555.
Здесь и далее приводятся предпочтительные таутомерные формы существования продуктов замещения