

Синтезы на основе 5,6-бис(диметиламино)аценафтилена
На правах рукописи
Мех Максим Александрович
СИНТЕЗЫ НА ОСНОВЕ
5,6-БИС(ДИМЕТИЛАМИНО)АЦЕНАФТИЛЕНА
02.00.03 – Органическая химия
АВТОРЕФЕРАТ
диссертации на соискание ученой степени
кандидата химических наук
Ростов-на-Дону – 2010
Работа выполнена на кафедре органической химии химического факультета Южного федерального университета.
Научный руководитель: доктор химических наук, профессор
Пожарский Александр Федорович
Официальные оппоненты: доктор химических наук, профессор
Межерицкий Валерий Владимирович
кандидат химических наук, с.н.с.
Бичеров Александр Викторович
Ведущая организация: Институт металлорганической химии
им. Г.А. Разуваева, РАН (г. Нижний Новгород)
Защита состоится “ 12 ” февраля 2010 года в 14.00 часов на заседании диссертационного совета Д 212.208.14 в НИИ физической и органической химии ЮФУ по адресу: 344090, г. Ростов-на-Дону, пр. Стачки, 194/2, конференц-зал.
С диссертацией можно ознакомиться в научной библиотеке Южного федерального университета (344006, г. Ростов-на-Дону, ул. Пушкинская, 148).
Автореферат разослан “____” января 2010 года.

Ученый секретарь
диссертационного совета,
доктор химических наук Морковник А.С.
ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ
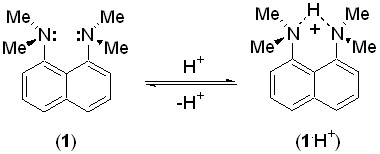
Актуальность работы. Нейтральные органические соединения с высокой основностью, сочетающейся с пониженной скоростью протонирования-депротонирования, получили название «протонные губки». Родоначальником этого ряда является 1,8-бис(диметиламино)нафталин (1) (pKa = 12.10, H2O[1] ), образующий хелатированный монокатион (1·Н+).
Из-за экранирования межазотного пространства метильными группами «протонные губки» обладают крайне низкой N-нуклеофильностью по отношению к электрофилам более крупным, чем протон. Благодаря этому, они часто используются в органическом синтезе, например, для селективного депротонирования субстратов, содержащих чувствительные к нуклеофилам группы. «Протонные губки» нашли применение для моделирования коротких низкобарьерных водородных связей, что весьма важно для понимания процессов протонного переноса в живых клетках (работа ферментативных систем). В последние годы предприняты попытки создания на основе «протонных губок» сенсоров и функциональных молекулярных устройств.
Объектом настоящего исследования стал 5,6-бис(диметиламино)-аценафтилен (2), являющийся аценафтиленовым аналогом соединения 1. Диамин 2 был впервые получен на кафедре органической химии ЮФУ дегидрированием аценафтена 3 (V.A. Ozeryanskii et al., J. Org. Chem., 2000, 65, 7707). Тогда же была продемонстрирована легкость взаимоперехода 2 3, открывающая путь к простому регулированию основности «протонных губок» (сразу на четыре порядка), что может быть полезно при конструировании молекулярных машин, откликающихся на изменения рН среды.
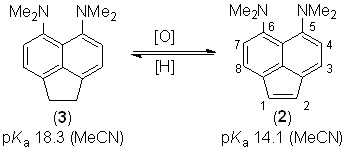
Нам казалось интересным одновременное присутствие в молекуле 2 высокоосновного центра и двойной связи С(1)=С(2). Уже в самом аценафтилене эта связь существенно обособлена от нафталиновой -системы и, по существу, определяет его реакционную способность. Поэтому априори соединение 2 можно было одновременно рассматривать как активированный алкен, «протонную губку» и даже «расширенный» енамин. В совокупности это позволяло ожидать у него проявления свойств, не похожих на свойства аценафтилена или родоначальной «протонной губки» 1.
Цели работы:
1) изучить реакционную способность 5,6-бис(диметиламино)аценафтилена по отношению к различным электрофилам, включая галогенирующие, формилирующие и нитрующие агенты, выяснить факторы, влияющие на направление этих реакций;
2) ввести полученные галогениды в реакцию Соногаширы с целью синтеза этинилпроизводных аценафтиленовой «протонной губки»;
3) разработать методы синтеза конденсированных гетеросистем на основе 5,6-бис(диметиламино)аценафтилена;
4) исследовать физико-химические свойства полученных соединений.
Научная новизна и практическая значимость. Установлено, что в отличие от более основных «протонных губок» 1 и 3, 5,6-бис(диметиламино)-аценафтилен 2 подвергается кислотному H/D-обмену в среде CF3CO2D. Реакция протекает через монопротонированную форму основания и в ней участвуют как этиленовые протоны Н-1,2, так и протоны нафталинового ядра. Соотношение активностей протонов Н-1(2) : Н-3(8) : Н-4(7) в катионе 5,6-бис(диметиламино)-аценафтилена составляет 3 : 1.3 : 1.
Показано, что пери-диметиламиногруппы активируют аценафтиленовую систему в такой степени, что активность нафталинового фрагмента и 1,2-двойной связи становятся соизмеримыми. Получены галогениды, альдегиды, нитро- и сульфонилпроизводные аценафтиленовой протонной губки, в которых функциональные группы расположены как при двойной связи, так и в нафталиновом кольце.
При взаимодействии соединения 2 со свободными галогенами первоначально образующиеся продукты присоединения по связи С(1)=С(2) из-за присутствия в молекуле высоко основного фрагмента быстро подвергаются Е2-элиминированию, образуя 1-галоген- или 1,2-дигалогенпроизводные с хорошим выходом, что ранее не наблюдалось в ряду аценафтилена.
Проведенный рентгеноструктурный анализ в сочетании с измерением основности выявил наличие в 4,7-дигалоген-5,6-бис(диметиламино)-аценафтиленах «эффекта поддержки».
Продукты замещения, содержащие в пятичленном цикле сильные –М-заместители (NO2, CHO, ArSO2) представляют собой ярко выраженные «пуш-пульные» системы с эффективным сквозным сопряжением, обладающие глубокой окраской и сольватохромией.
Синтезирован ряд конденсированных гетероциклических систем на основе 5,6-бис(диметиламино)аценафтилена, а также ацетиленовые соединения данного ряда.
Апробация работы и публикации. Основные результаты работы представлены на Всероссийской научной конференции «Современные проблемы органической химии», посвященной 100-летию со дня рождения академика Н.Н. Ворожцова, Новосибирск, 2007 г и на IX Международном семинаре по магнитному резонансу (Спектроскопия, Томография и Экология), Ростов-на-Дону, 2008 г.
По материалам диссертации опубликовано 3 статьи и 2 тезисов докладов.
Объем и структура работы. Диссертация состоит из введения, трех глав, выводов, списка цитированной литературы и приложения. Первая глава представляет собой литературный обзор, посвященный методам получения и свойствам аценафтиленов. Вторая глава содержит результаты собственных исследований автора. Третья глава – экспериментальная часть.
Работа изложена на 101 странице машинописного текста, содержит 35 схем, 52 графических уравнения, 26 таблиц и 14 рисунков. Библиография насчитывает 159 наименований.
Исследование выполнено при частичной финансовой поддержке РФФИ (проекты №№ 05-03-32110, 08-03-00028).
ОСНОВНОЕ СОДЕРЖАНИЕ РАБОТЫ
1. Взаимодействие 5,6-бис(диметиламино)аценафтилена с электрофилами
Кислотный дейтерообмен. Н/D-Обмен проводился в дейтеротрифторуксусной кислоте, в которой соединение 2 хорошо растворимо. За ходом процесса следили с помощью спектроскопии ЯМР 1Н. Реакция протекает с удобной для измерений скоростью при 65 оС. Легкость обмена протонов Н-1,2 почти в три раза выше, чем протонов нафталинового ядра (рис. 1). При этом активность положений 3 и 8 в последнем несколько больше, чем положений 4 и 7. В целом соотношение активностей протонов Н-1(2) : Н-3(8) : Н-4(7) = 2.96 : 1.33 : 1. Сам факт, что обмен облегчается по мере удаления реакционного центра от NMe2 групп, свидетельствует о том, что реакция протекает через протонированную форму основания 2.
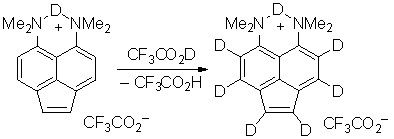
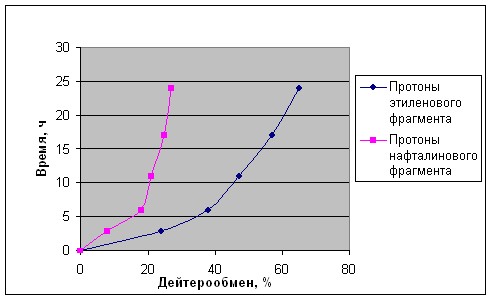
Рис. 1. Ход кислотного H/D обмена протонов нафталинового кольца и протонов связи С(1)=С(2) (CF3CO2D, 65 oC)
Галогенирование. Взаимодействие 5,6-бис(диметиламино)аценафтилена (2) с бромом в сравнении с незамещенным аценафтиленом и большинством его производных протекает весьма специфично. Если последние ведут себя как типичные алкены, образуя смесь цис- и транс-1,2-дибромаценафтенов, то соединение 2 при действии 1 экв. брома (в CCl4, бензоле или уксусной кислоте) дает смесь моно- и дибромаценафтиленов 4 и 5 с суммарным выходом 50–60% и заметным преобладанием монобромида; одновременно регенерируется около 30–50% исходного соединения. Очевидно, что первоначально образующийся аддукт 6 быстро дегидробромируется молекулой 2 с образованием монобромида 4 и гидробромида 2·HBr. Повторное присоединение брома к 4 и дегидробромирование аддукта 7 дают дибромид 5. Можно предположить, что соль 2·HBr реагирует с бромом медленнее, чем основание 2, поскольку в катионе двойная связь С=С не активирована донорным эффектом NMe2-групп.
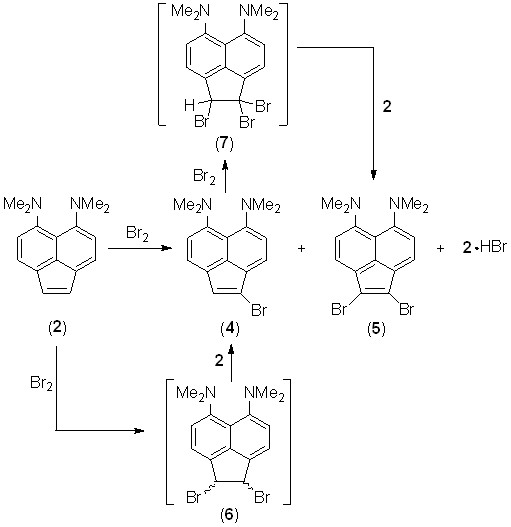
Избежать дегидробромирования дибромаценафтена 6 удается, если вводить в реакцию соль «протонной губки» 2. Так, при действии 1 экв. брома на соль 2·HClO4 в ацетонитриле с последующей обработкой реакционной смеси водным раствором Na2CO3 монобромид 4 получается с почти количественным выходом. В качестве промежуточного продукта здесь выступает соль 6·HClO4, которую можно выделить, удалив при комнатной температуре ацетонитрил из реакционной смеси до обработки содой. В спектре ЯМР 1Н (рис. 2) соединения 6·HClO4 имеется два дублета (JNH,NMe = 2.6 Гц) одинаковой интенсивности с химическими сдвигами 3.13 и 3.15 м.д., соответственно, что свидетельствует о наличии в катионе 6·H+ двух пар магнитно-неэквивалентных N-метильных групп. Действительно, как в цис-, так и в транс-дибромиде 6·HClO4 две CH3-группы противостоят атомам брома, а две другие – атомам водорода в позициях 1 и 2. К сожалению, исходя из этих данных, нельзя сделать выбор в пользу того или иного изомера и, таким образом, судить о стереохимии присоединения галогена.

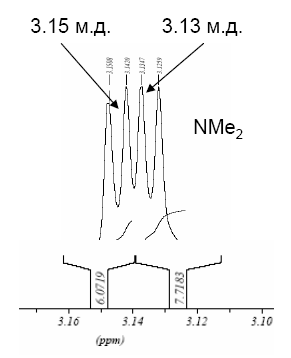
Рис. 2. Фрагмент спектра ЯМР 1H соединения 6·HClO4 (300 МГц, CD3CN)
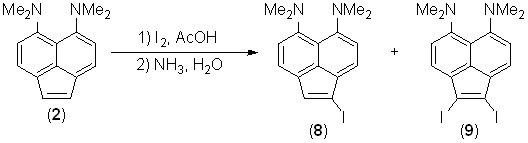
Взаимодействие йода с аценафтиленом 2 в уксусной кислоте при комнатной температуре протекает аналогично бромированию и дает йодпроизводные 8 и 9 – весьма лабильные, особенно при хроматографировании, вещества. Выходы соединений 8 и 9 с 1 экв. йода – 36 и 15%, соответственно. С 2 экв. йода монойодид 8 образуется лишь в следовых количествах, тогда как выход дийодида 9 достигает 40%.
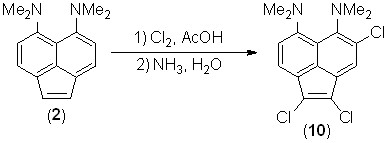
При действии на соединение 2 раствора молекулярного хлора в хлороформе в интервале температур от 25 до –15 оС наблюдается полное осмоление. Однако при замене хлороформа на уксусную кислоту был получен красный трихлорид 10 с выходом 5–16% (в зависимости от количества хлора). Других продуктов выделить не удалось.
Хлорирование диамина 2 N-хлорсукцинимидом (NCS) в хлороформе или ДМФА (при –15 оС) селективно протекает по орто-положениям к диметиламиногруппам. При этом, в зависимости от взятого количества NCS, можно получить с высоким выходом как монохлорид 11, так и дихлорид 12. Возможность селективного орто-монохлорирования говорит о меньшей скорости вхождения второго атома хлора. Кроме того, сам факт протекания реакции по нафталиновому ядру молекулы 2 принципиально важен, поскольку для самого аценафтилена примеров подобных реакций практически нет.
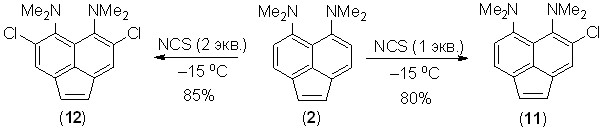
Дихлорпроизводное 12 может быть получено и без растворителя путем растирания исходного соединения 2 с 2 экв. NCS при комнатной температуре (выход 67%).
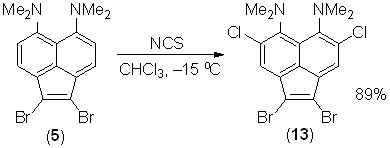
4,7-Дихлорид 12 не подвергается дальнейшему хлорированию NCS или бромированию NBS при кипячении в хлороформе. Очевидно, это объясняется недостаточной активацией кольца из-за сильного поворота NМe2-групп в совокупности с электроноакцепторным эффектом атомов хлора. В то же время, 1,2-дибромид 5, в котором сопряжение диметиламиногрупп с -системой существенно не нарушено, реагирует с 2 экв. NCS в хлороформе уже при –15 оС, образуя с высоким выходом смешанный тетрагалогенид 13.
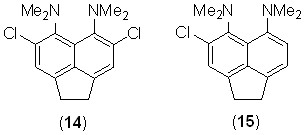
Каталитическое гидрирование (Pd/C, H2) дихлорида 12 в этаноле приводит к восстановлению двойной связи и образованию с количественным выходом дихлораценафтена 14. Напротив, монохлорид 11 как в виде основания, так и перхлората, в тех же условиях подвергается не только гидрированию, но и протодехлорированию, образуя в качестве единственного продукта аценафтеновую «протонную губку» 3, но не соединение 15. Аналогичные результаты были получены при проведении реакции в метаноле, этилацетате или ТГФ. Причина повышенной неустойчивости связи C–Cl в этом случае не ясна.
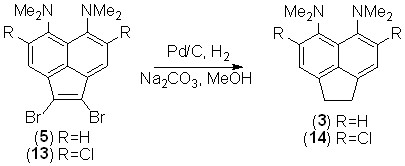
При каталитическом восстановлении дибромидов 5, 13 в присутствии карбоната натрия (для связывания выделяющегося HBr) наряду с гидрированием двойной связи наблюдается дебромирование, в результате чего с количественным выходом образуются аценафтены 3 и 14, соответственно.
Действие на 5,6-бис(диметиламино)аценафтилен (2) NBS в хлороформе при –15 оС дает сложную смесь веществ, которую не удалось разделить. Замена CHCl3 на ДМФА делает процесс более контролируемым и при использовании 1 экв. NBS позволяет получить с небольшим выходом 1-бром- 4 и 1,2-дибром-производные 5. С 2 экв. NBS дибромид 5 становится единственным продуктом реакции (12%); при дальнейшем увеличении количества бромирующего агента третий и четвертый атомы брома вступают в орто-положения к NMe2-группам, но одна из них при этом деметилируется. Процесс деметилирования, вероятно, носит окислительный характер и протекает с участием катион-радикалов. Выходы полибромидов 16, 17 – 10–24%. Понижение температуры реакционной смеси от –15 до –57 оС практически не влияет на результат.
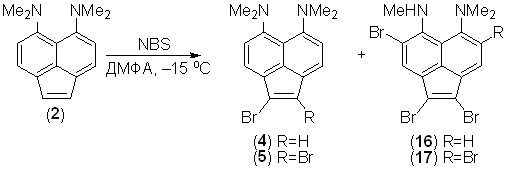
Для трибромида 16 был проведен рентгеноструктурный (РС) анализ, который доказал положение атома брома в нафталиновом фрагменте молекулы и, кроме того, выявил наличие слабой внутримолекулярной водородной связи N–H···N между группами NMe2 и NHMe: для двух кристаллографически независимых молекул расстояние N···N равно 2.75 и 2.76, NHN – 125 и 129o, соответственно (рис. 3).
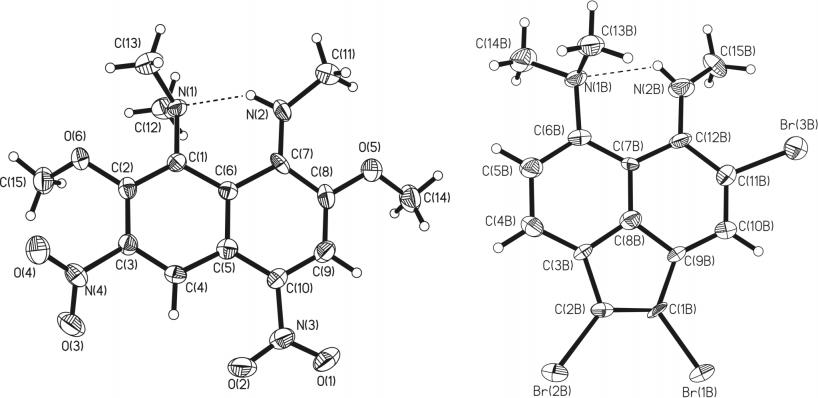
Рис. 3. Общий вид одной из двух кристаллографически независимых молекул трибромида 16 (293 K). Тепловые эллипсоиды показаны на уровне 30%
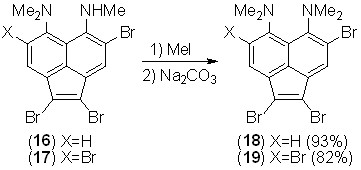
Длительное кипячение N-триметилпроизводных 16 и 17 с избытком йодметана с последующей нейтрализацией смеси приводит к образованию с высоким выходом соединений 18 и 19.
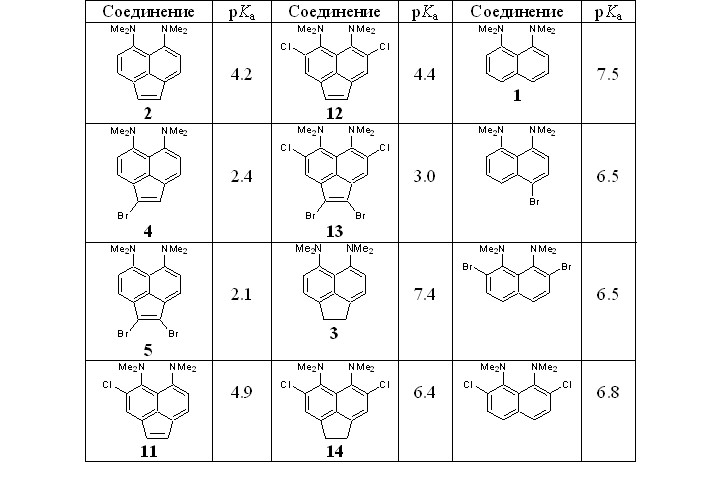
При помощи метода ЯМР 1Н посредством конкурентного протонирования нами были измерены величины pKa некоторых синтезированных галогенидов и модельных оснований в ДМСО (табл. 1). Установлено, что атом брома в положении 1 аценафтиленовой «протонной губки» уменьшает основность на 1.8 ед. рKа (соединения 2 и 4), при введении второго брома в положение 2 падение основности существенно меньше (соединение 5).
Влияние атомов хлора в положениях 4 и 7 менее однозначно (соединения 11 и 12). Если первый заместитель вызывает рост основности на 0.7 ед. pKa, то при вхождении второго основность падает, хотя и остается чуть более высокой, чем у незамещенного субстрата 2. Основность смешанного тетрагалогенида 13 отражает наложение указанных тенденций. Интересно, что в нафталиновой 1 и аценафтеновой «протонных губках» 3 влияние атомов хлора, находящихся в орто-положениях к группам NMe2, совершенно иное и приводит к уменьшению основности на 0.7 и 1.0 pKa, соответственно.
Таблица 1. Константы основности, pKa, некоторых синтезированных галогенпроизводных и модельных соединений (ДМСО, 22 oC)
Как показали РС исследования дихлораценафтилена 12 и его моноперхлората, специфичное влияние атомов хлора проявляется в сильном сближении NMe2-групп в пространстве (до 2.87 ) и их значительном развороте относительно плоскости ароматической системы (67о в основании 12 и 88о в соли 12·HClO4), что облегчает последующее протонирование.
Формилирование. Мы установили, что соединение 2 формилируется по методу Вильсмайера в толуоле в интервале температур от –15 до 25 оС, давая смесь моноальдегида 20 и диальдегидов 21, 22. Реакция проводилась в присутствии большого избытка ДМФА, играющего роль не только реагента, но и сорастворителя. Результаты опытов существенно зависели от количества взятого хлороксида фосфора. При использовании 1–3 экв. POCl3 получалось 6–9% моноальдегида и лишь следы диальдегидов. С 5 экв. POCl3 выход моноальдегида изменялся мало, но образовывалось соизмеримое количество 1,2- и 1,7-диальдегидов. Суммарный выход альдегидов в лучших опытах достигал 20%, около половины исходного соединения регенерировалось. Несомненно, это обусловлено выделением в ходе реакции хлористого водорода, который связывался соединением 2, превращая его в инертный к формилированию катион. Ранее было найдено (В.А. Озерянский и др., Изв. АН, Сер. хим., 2004, 388–397), что в подобных случаях реакцию можно довести до конца, прибавляя в смесь намного более сильное и малонуклеофильное основание, например, 1,8-бис(диметиламино)-2,7-диметоксинафталин (рKа = 16.1). Действительно, в присутствии 1 экв. этого вещества суммарный выход альдегидов возрос до 40%. Интересно, что в отличие от соединения 2, аценафтеновая «протонная губка» 3 не реагирует с комплексом Вильсмайера даже при нагревании до 80 оС в течение часа.
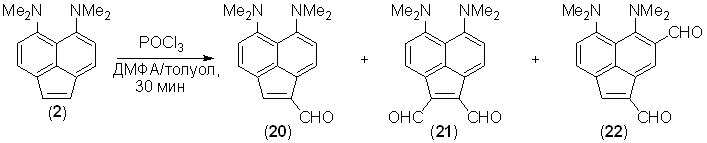
Диальдегид 21 представляет собой темно-фиолетовое кристаллическое вещество с макс 550 нм (lg 4.39, CH2Cl2), обладает металлическим блеском и внешне похоже на перманганат калия. Эти признаки, очевидно, отражают весьма эффективное «сквозное» сопряжение групп CHO и NMe2. В этом отношении соединение 21 превосходит пери-диальдегид 27 (макс 444 нм, lg 4.11, CHCl3 [V.A.Ozeryanskii et al., Eur. J. Org. Chem., 2009, 1241]). Оба диальдегида заметно растворимы в воде, что объясняется их высоким дипольным моментом.
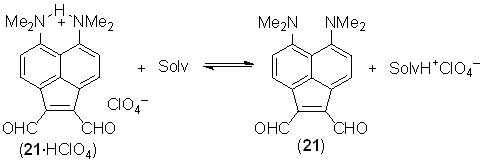
Другим результатом сопряжения в диальдегиде 21 является его пониженная основность, из-за чего протонные соли, например 21·HClO4, имеют зеленоватый цвет и депротонируются в достаточно основных растворителях, проявляя положительную сольватохромию в весьма широком спектральном диапазоне (>210 нм, рис. 4).

Рис. 4. Сольватохромия перхлората 21·HClO4 (каждая ампула содержит 2 мг соли и 4 мл растворителя)
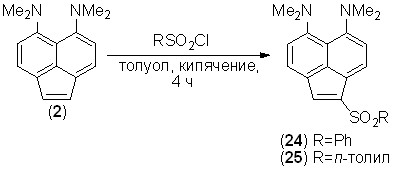
Арилсульфонирование и нитрование. При действии на аценафтилен 2 1 экв. бензол- или п-толуолсульфохлорида в кипящем толуоле с выходом до 30% образуются сульфоны 24 и 25. Часть исходного соединения 2, как и при формилировании, протонируется образующимся в ходе реакции хлороводородом и не подвергается сульфонированию. Реакция затрагивает исключительно положение 1.
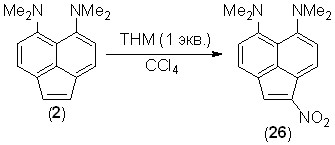
Мы нашли, что обработка соединения 2 1 экв. HNO3 в серной или уксусной кислоте, а также в ацетонитриле в интервале температур от комнатной до –15 оС приводит к полному осмолению. Действие на 2 диоксида азота (1 экв.) в ССl4 при –18 оС также не дала положительного результата. Тем не менее, при обработке соединения 2 1 экв. тетранитрометана (ТНМ) в CCl4 при –7…–14 оС было получено 1-нитропроизводное 26 с выходом 13%; другие продукты выделить не удалось. Использование 2 экв. ТНМ также привело к полному осмолению. Соединение 26 представляет собой темно-фиолетовое кристаллическое вещество, обладающее металлическим блеском, но, в отличие от диальдегида 21, не окрашивающее воду.
2. Синтез 1-этинил- и 1,2-диэтинилпроизводных
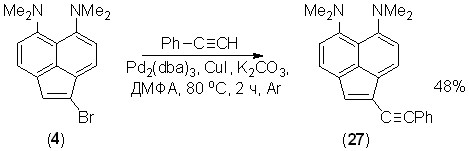
Синтезированные нами бромиды аценафтиленовой «протонной губки» были использованы для получения ее этинилпроизводных. Так, соединения 4, 5 и 13 дают с фенилацетиленом в условиях реакции Соногашира ацетилены 27, 28 и 29; при этом атомы хлора в случае полигалогенида 13 не затрагиваются.

Строение диацетилена 28 подтверждено РС анализом (рис. 5). Расстояние N(1)···N(2) составляет 2.981 против 2.955 для молекулы 2; примерно такая же разница наблюдается и в длинах связи C(1)=C(2) для соединений 28 и 2: 1.378 и 1.339, соответственно. Эти различия можно рассматривать как результат достаточно эффективного сопряжения фенилэтинильных заместителей с NМe2-группами. Двугранные углы между плоскостями аценафтиленовой системы и бензольных колец С(19)С(20)…С(24) и С(27)С(28)…С(32) равны 171.2 и 120.4°, соответственно.
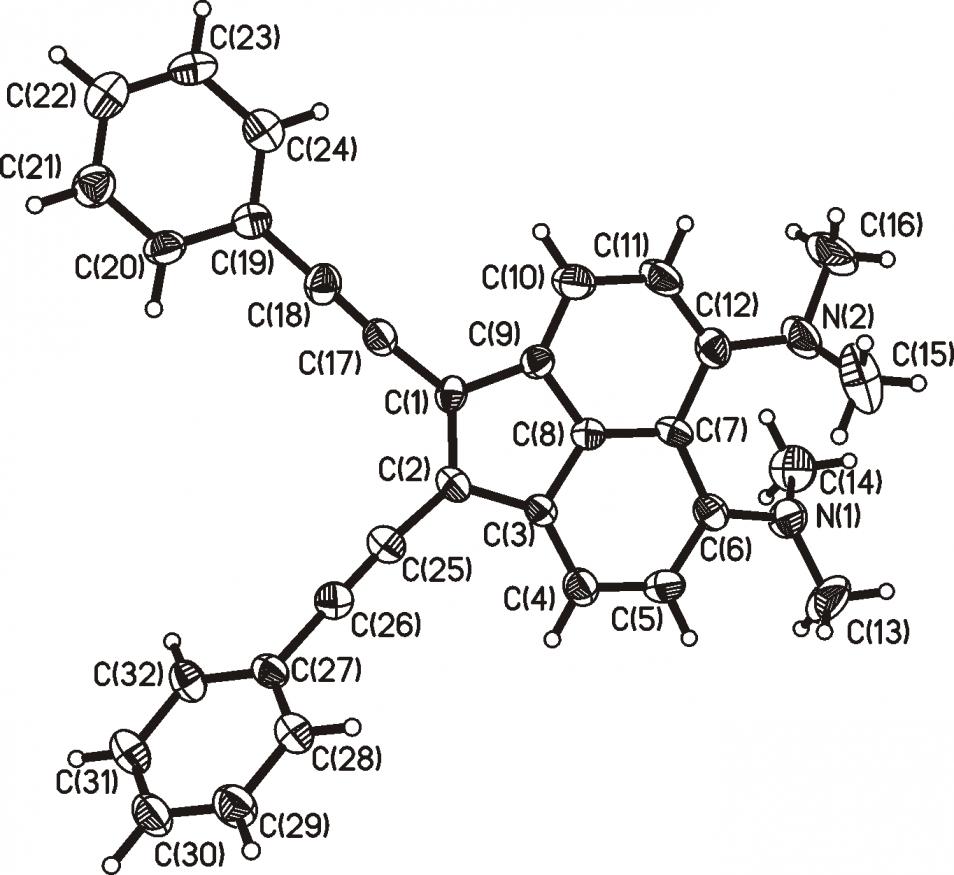
Рис. 5. Молекулярная структура соединения 28 (293 K). Тепловые эллипсоиды показаны на уровне 30%
Аналогичным образом реакция бромидов 4, 5 с триметилсилилацетиленом в присутствии Pd2(dba)3, CuI и Et3N привела к образованию триметилсилилпроизводных 30 и 31. Десилилированием последнего получен диацетилен 32 – весьма лабильное красное масло, быстро темнеющее на воздухе.

3. Синтез конденсированных гетероциклических систем
Недавно в нашей лаборатории было показано (А.Ф. Пожарский и др., Изв. АН. Сер. хим., 2003, 206), что соединение 2 проявляет свойства активного диенофила, вступая в реакцию [4+2]-циклоприсоединения с обращенными электронными требованиями. Так, взаимодействие 2 с симм-тетразинами сопровождается выделением азота и, после окисления, дает соответствующие пиридазины. Мы провели аналогичные превращения с хлоридами 11 и 12. При действии на них 1 экв. 3,6-дифенилтетразина 33 (толуол, 110 оС, 15 ч) с высоким выходом образуются пиридазины 34, 35. Действие на пиридазин 36 NCS также дает дихлорид 35, но по сравнению с 2 реакция хлорирования протекает труднее. Остановить превращение на стадии образования монохлорида 34 не удается.

Как и следовало ожидать, 5,6-динитроаценафтилен (37), этиленовый фрагмент которого пассивирован нитрогруппами, вступает в циклоприсоединение заметно труднее. Его взаимодействие с 3,6-дифенилтетразином (о-ксилол, 140 оС, 48 ч) дает пиридазин 38 лишь с выходом 21% (структура подтверждена РС анализом). Мы полагали, что динитропиридазин 38, в молекуле которого энергия НСМО, очевидно, должна быть понижена по сравнению с 36, будет реагировать с 2, образуя «пуш-пульное» соединение 39. К сожалению, сложную смесь, образовавшуюся при сплавлении (180 оС) соединения 38 с аценафтиленом 2, разделить не удалось, а в более мягких условиях реакция не протекала.
Обработка диазафлуорантена 40, синтезированного нами из 2 и 3,6-ди(метоксикарбонил)-симм-тетразина, цинком в уксусной кислоте по известной реакции восстановительного сужения пиридазинового цикла привела с выходом 57% к 3,4-бис(диметиламино)-7,9-ди(метоксикарбонил)аценафто[1,2-c]пирролу (41) – первому пиррол-содержащему производному «протонной губки». Это соединение представляет собой желтое кристаллическое вещество, хорошо растворимое в обычных органических растворителях. В его спектре ЯМР 1Н в CDCl3 протон NH проявляется при 9.31 м.д., что позволяет исключить образование в растворе бетаина с протоном, хелатированным между NMe2-группами (величина для NH-протона в таких катионах обычно лежит в интервале 16–19 м.д.). Действительно, измеренная нами константа кислотности пиррола 41 в ДМСО-d6 равна pKa = 12.5, что явно недостаточно для перескока протона NH.
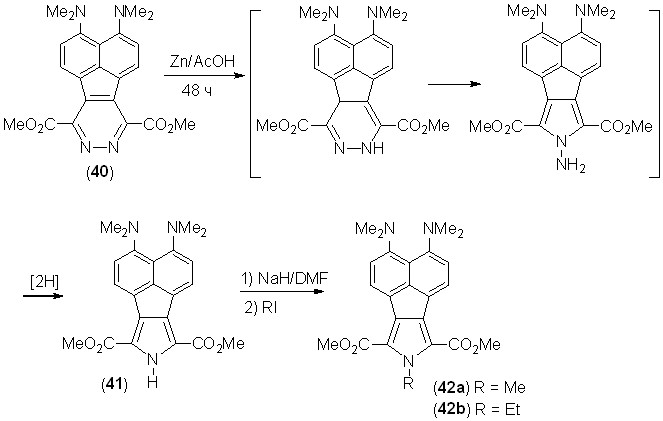
Ионизация соединения 41 1 экв. гидрида натрия в ДМФА с последующей обработкой реакционной смеси MeI или EtI дает N-алкилпирролы 42a и 42b с выходом 56 и 72%, соответственно.
Пиррол 41 подвергается хлорированию NCS в хлороформе с образованием дихлорида 43. Реакция даже при –15 оС сопровождается сильным смолообразованием, выход дихлорида 43 не превышает 11%.
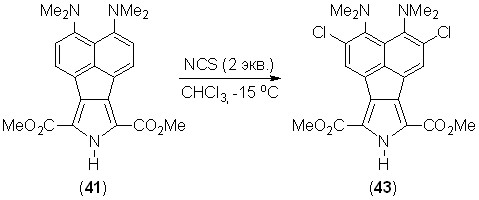
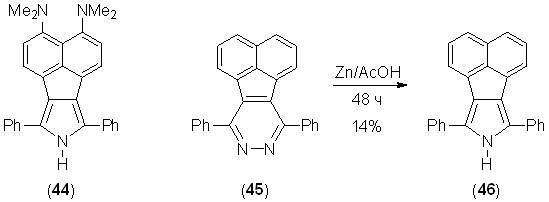
Попытка осуществить синтез пиррола 44 из соответствующего пиридазина не увенчалась успехом. Полученное желтое флуоресцирующее вещество быстро разлагалось в процессе очистки. По-видимому, соединение 44 нестабильно из-за высокой -избыточности. В самом деле, полученный нами из пиридазина 45 и не содержащий донорных NMe2-групп пиррол 46 (бледно-желтое флуоресцирующее вещество) оказался вполне устойчивым в обычных условиях.
ВЫВОДЫ
- Показано, что в отличие от 1,8-бис(диметиламино)нафталина («протонной губки»), 5,6-бис(диметиламино)аценафтилен подвергается H/D-обмену в CF3CO2D, причем легче всего обмениваются мостиковые протоны Н-1,2. Порядок изменения активности протонов Н-1(2) : Н-3(8) : Н-4(7), составляюший 2.96 : 1.33 : 1, свидетельствует о том, что в реакции участвует катион 5,6-бис(диметиламино)аценафтилена.
- Исследовано галогенирование 5,6-бис(диметиламино)аценафтилена. При использовании N-галогенсукцинимидов направление реакции определяется главным образом размером галогена и полярностью растворителя, в зависимости от чего можно направлять процесс в сторону 1(2) или 4(7)-(ди)галогенпроизводных. Реакция 5,6-бис(диметиламино)аценафтилена с молекулярным бромом или йодом дает продукт присоединения по связи С(1)=С(2), который из-за высокой основности субстрата быстро подвергается Е2-элиминированию, образуя 1-галоген- или 1,2-дигалогенпроизводные с хорошим выходом. В реакции с хлором удается выделить только 1,4,7-трихлорид.
- С помощью конкурентного транспротонирования в ДМСО определены значения pKa полученных галогенидов аценафтеновых и аценафтиленовых «протонных губок». Введение атомов хлора в положения 4(7) ведет к некоторому увеличению основности, тогда как атомы брома в положениях 1(2) значительно снижают величины pKa. Как показали рентгеноструктурные исследования 5,6-бис(диметиламино)-4,7-дихлораценафтилена и его моно-перхлората, влияние атомов хлора проявляется в сильном сближении диметиламиногрупп («эффект поддержки») и их значительном развороте относительно плоскости ароматической системы, что облегчает протонирование.
- Показано, что 5,6-бис(диметиламино)аценафтилен в мягких условиях подвергается арилсульфонированию и нитрованию, образуя с умеренным выходом продукты замещения по положению 1. В отличие от этого, формилирование по Вильсмайеру, наряду с 1-формил- и 1,2-диформилпроизводными, дает некоторое количество 1,7-дикарбальдегида, что, как и галогенирование, демонстрирует сравнимую реакционную способность этиленового и нафталинового фрагментов в непротонированном субстрате.
- Показано, что пери-диметиламиногруппы активируют аценафтиленовую систему в такой степени, что активность нафталинового фрагмента и 1,2-двойной связи становятся соизмеримыми. Это контрастирует с поведением других аценафтиленов, где объектом электрофильной атаки практически всегда является 1,2-двойная связь.
- На основе 1-бром- и 1,2-дибром-5,6-бис(диметиламино)аценафтиленов с помощью реакции Соногаширы с хорошим выходом получены моно- и ди(этинил)производные.
- Синтезирован ряд производных 5,6-бис(диметиламино)аценафтилена с конденсированными пиридазиновыми и пиррольными кольцами в положениях 1,2. Полученные гетероциклы представляют интерес для построения на их основе порфиринсодержащих систем с высокоосновными фрагментами.
ОСНОВНОЕ СОДЕРЖАНИЕ ДИССЕРТАЦИИ ОПУБЛИКОВАНО В СЛЕДУЮЩИХ РАБОТАХ:
- Кажева О.Н., Шилов Г.В., Дьяченко О.А., Мех М.А., Сорокин В.И., Озерянский В.А., Пожарский А.Ф. 1-Диметиламино-2,7-диметокси-8-метиламино-3,5-динитронафталин и 1,2,4-трибром-6-диметиламино-5-метиламиноаценафтилен: первые примеры сжатия/растяжения N–H…N водородной связи в нейтральных 1,8-диаминонафталинах // Изв. АН, Сер. хим., 2005, № 11, 2414–2417.
- Mekh M.A., Ozeryanskii V.A., Pozharskii A.F. 5,6-Bis(dimethylamino)acenaphthylene as an activated alkene and 'proton sponge' in halogenation reactions // Tetrahedron, 2006, 62, № 52, 12288–12296.
- Мех М.А., Озерянский В.А., Пожарский А.Ф. Реакционная способность 5,6-бис(диметиламино)аценафтилена // Материалы Всероссийской научной конференции "Современные проблемы органической химии", посвященной 100-летию со дня рождения академика Н.Н. Ворожцова, Новосибирск, Июнь 5–9, 2007, 88.
- Мех М.А., Пожарский А.Ф., Озерянский В.А. – Сравнительная реакционная способность 1,2-двойной связи в аценафтилене и 5,6-бис(диметиламино)аценафтилене // Материалы IX Международного семинара по магнитному резонансу (спектроскопия, томография и экология), Ростов-на-Дону, Сентябрь 15–20, 2008, 160.
- Mekh M.A., Pozharskii A.F., Ozeryanskii V.A. // Electrophilic substitution in 5,6-bis(dimethylamino)acenaphthylene as a route to push-pull proton sponges, Polish J. Chem., 2009, 83, № 9, 1609–1621.
[1] Здесь и далее под величинами pKa азотистых оснований понимаются величины pKBH+, характеризующие диссоциацию сопряженных кислот.