

Клиническое и молекулярно-цитогенетическое исследование синдрома вильямса
На правах рукописи
Прокофьева Алёна Дмитриевна
КЛИНИЧЕСКОЕ И МОЛЕКУЛЯРНО-ЦИТОГЕНЕТИЧЕСКОЕ
ИССЛЕДОВАНИЕ СИНДРОМА ВИЛЬЯМСА
Специальность: 03.00.15 — генетика
14.00.09 — педиатрия
АВТОРЕФЕРАТ
диссертации на соискание
учёной степени кандидата медицинских наук
Санкт-Петербург - 2006
Работа выполнена на кафедре медицинской генетики Государственного образовательного учреждения дополнительного профессионального образования «Санкт-Петербургская медицинская академия последипломного образования Федерального агентства по здравоохранению и социальному развитию», в Санкт-Петербургском Государственном учреждении здравоохранения «Диагностический центр (медико-генетический)» Комитета по здравоохранению Правительства Санкт-Петербурга, в лаборатории Стабильности хромосом и клеточной инженерии Института цитологии Российской Академии Наук.
.
Научные руководители:
- доктор биологических наук, профессор Н.В.Томилин,
- доктор медицинских наук О.П. Романенко
Официальные оппоненты:
доктор медицинских наук, профессор А. Н. Петрин
кандидат медицинских наук, доцент А.Н.Прытков
Ведущее учреждение:
НИИ экспериментальной медицины РАМН
Защита состоится 25 сентября 2006 г., в 11-00 на заседании Диссертационного совета Д 001.016.01 при Государственном учреждении «Медико-генетический научный центр Российской Академии медицинских наук» по адресу: г. Москва, ул. Москворечье, 1 …
С диссертацией можно ознакомиться в библиотеке ГУ МГНЦ РАМН
Автореферат разослан 24/У111-2006
Учёный секретарь Диссертационного совета д.б.н., профессор Л.Ф. Курило.
ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ
Актуальность исследования: В структуре задержки психического развития у детей синдром Вильямса (СВ) является одной из распространённых, нозологически самостоятельных форм умственной отсталости и по значимости занимает третье место после синдрома Дауна и фенилкетонурии (Г.С. Маринчева, В.И. Гаврилов, 1988). СВ был впервые описан в середине ХХ века Fanconi с соавт. (1952), и до конца прошлого столетия его диагностика проводилась на основании клинических данных, поскольку у больных как правило определяется нормальный кариотип (A.K. Ewart и др., 1993; E. Nickerson и др., 1995; L.A. Perez Jurado и др., 1996). После стремительного развития методов молекулярной генетики, открытия критического района генома 7q11.23 и описания гемизиготной делеции гена эластина в этом районе при СВ (A.K. Ewart и др., 1993) стала доступной диагностика этого заболевания на молекулярном уровне. В настоящее время в различных странах для подтверждения клинического диагноза СВ используется молекулярное и молекулярно-цитогенетическое исследование.
Для СВ характерно умеренное снижение интеллекта, что, в сочетании с относительно сохранным речевым развитием и часто выраженными музыкальными способностями, создаёт благоприятную почву для социальной реабилитации больных (Г.С. Маринчева, В.И. Гаврилов, 1988). В последнее время большое внимание уделяется полиорганному характеру поражения организма при данном заболевании, что также требуется учитывать при планировании реабилитационных мероприятий для конкретного пациента (K. Metcalfe, 1999).
Данные о частоте СВ в Санкт-Петербурге при изучении литературы не найдены. Молекулярно-цитогенетическая диагностика этого синдрома в России проводится в Государственном Учреждении «Медико-генетический научный центр Российской Академии Медицинских Наук» (Москва).
Цель работы: На основании комплексного — клинического и молекулярно-цитогенетического — подхода изучить СВ у детей и обосновать рекомендации по улучшению диагностики данного заболевания.
Задачи исследования:
- Изучить клиническую картину у больных с СВ: выявить характерные симптомы заболевания и дополнительные особенности.
- На основе предоставленных продуктов первичной амплификации искусственных дрожжевых хромосом создать в лабораторных условиях ДНК-зонды, пригодные для проведения молекулярно-цитогенетическое исследования; произвести оценку специфичности и чувствительности полученных ДНК-зондов, осуществив молекулярно-цитогенетическое исследование на контрольном материале (хромосомных препаратах, полученных от лиц без клинической симптоматики СВ).
- Провести молекулярно-цитогенетическое исследование на хромосомных препаратах, полученных от больных с СВ, с применением созданных ДНК-зондов.
- Сопоставить клиническую картину с данными молекулярно-цитогенетического исследования при СВ.
Научная новизна: Впервые в России показано, что для подтверждения клинического диагноза синдрома Вильямса можно и нужно использовать высокоточного современного метода FISH с несколькими локус-специфичными ДНК-зондами, целиком перекрывающими критический для СВ район 7 хромосомы (7q11.2). Выявлены варианты делеций: STS-маркера D7S489B, STS-маркера D7S672 и обоих исследованных STS-маркеров (D7S489B и D7S672).
Впервые описан феномен дополнительной микроделеции в прицентромерной области 7р11.1-7р11.2 при наличии делеции STS-маркера D7S489B в критическом для СВ районе 7q11.2, а также при дизиготном состоянии локусов D7S494 и D7S672.
Впервые проведено сопоставление типичных клинических проявлений СВ с делециями в области STS-маркеров: D7S489B (7q11.2), D7S672 (7q11.2), D7S494 (7p11.1-7p11.2) у больных с СВ, не являющихся родственниками. Впервые сопоставлены дополнительно выявленные аномалии и особенности фенотипа у больных с СВ, не являющихся родственниками, и делеции в области STS-маркеров: D7S489B (7q11.2), D7S672 (7q11.2), D7S494 (7p11.1-7p11.2). Впервые установлено сочетание поздней гиперкальцемии с одновременной делецией двух STS-локусов D7S489B (7q11.2) и D7S494 (7p11.1-7p11.2). Впервые выявлена гипокальцемия при синдроме Вильямса при различных вариантах делеций исследованных STS-маркеров. Впервые установлена артериальная гипотензия (в сочетании с анамнестической гиперкальцемией) при наличии гемизиготных делеций в двух STS-локусах: D7S489B (7q11.2) и D7S494 (7p11.1-7p11.2).
Практическая значимость:
- Показаны индивидуальная вариабельность типичных симптомов, возможное наличие нетипичных пороков развития, микроаномалий и функциональных особенностей при СВ, что должно учитываться при клинической диагностике и диспансерном наблюдении этих больных.
- Продемонстрирована адекватность и необходимость молекулярно-цитогенетического исследования критического района 7 хромосомы при СВ с применением двух использованных ДНК-зондов 743 g06 (STS-маркер D7S489B) и 893 b12 (STS-маркер D7S672) для уточнения протяжённости делеции.
- Впервые установленный феномен делеции STS-маркера D7S494 в прицентромерной области 7р11.1-7р11.2 при наличии микроделеции в районе 7q11.2 может быть использован для дальнейшего изучения механизма возникновения микроделеции в критическом для СВ районе и разработки концепции медико-генетического консультирования семей с пробандами, страдающими СВ.
Внедрение в практику: Вариабельность клинической картины СВ учитывается врачами-генетиками Санкт-Петербургском Государственном учреждении здравоохранения «Диагностический центр (медико-генетический)» Комитета по здравоохранению Правительства Санкт-Петербурга (МГЦ) при исследовании фенотипа больных с подозрением на диагноз: синдром Вильямса.
Молекулярно-цитогенетический метод обследования используется для верификации клинического диагноза СВ у больных, проходящих обследование в МГЦ.
Разработанные рекомендации по диспансерному наблюдению больных с установленным диагнозом синдрома Вильямса используются врачами-генетиками МГЦ в клинической работе.
Данные, полученные в ходе исследования, используются при подготовке лекционного материала сотрудниками кафедры медицинской генетики Государственного образовательного учреждения дополнительного профессионального образования (ГОУ ДПО) «Санкт-Петербургская медицинская академия последипломного образования Федерального Агентства по Здравоохранению и социальному развитию».
Положения, выносимые на защиту:
1. Клинической картине синдрома Вильямса присуща индивидуальная вариабельность типичных симптомов. Характерный фенотип может включать дополнительные врождённые пороки, микроаномалии и функциональные особенности.
2. Уровни специфичности и чувствительности созданных ДНК-зондов являются достаточными для выявления делеции соответствующих STS-маркеров.
3. Молекулярно-цитогенетический метод исследования (флюоресцентная гибридизация in situ, на хромосомных препаратах) является основным и необходимым лабораторным способом подтверждения клинического диагноза синдрома Вильямса. Хромосомная микроделеция в критическом районе длинного плеча 7 хромосомы имеет варьирующую протяжённость. В некоторых случаях при синдроме Вильямса обнаруживается дополнительная делеция за пределами критического района, в прицентромерной области короткого плеча 7 хромосомы.
4. Выявленные варианты изменения структуры 7 хромосомы не имеют специфических клинических проявлений.
Апробация работы: Основные результаты диссертации отражены в 6 опубликованных работах и представлены на конференции “Лабораторная медицина — взгляд в будущее” (Санкт-Петербург, 2001), доложены на заседании кафедры медицинской генетики ГОУ ДПО «Санкт-Петербургская медицинская академия последипломного образования Федерального агентства по здравоохранению и социальному развитию» (Санкт-Петербург, 2002), Международной конференции “Врачи мира — пациентам” (Санкт-Петербург, 2003), на семинарах Государственного учреждения «Медико-генетический научный центр РАМН» (Москва, 2004, 2006).
Публикации: По материалам диссертации опубликовано 6 печатных работ, в том числе 2 в центральных научных журналах.
Объём и структура работы: Диссертационная работа состоит из введения, обзора литературы, главы с описанием материалов и методов работы, трёх глав собственных результатов и их обсуждения, заключения, выводов, практических рекомендаций, списка литературы, приложений. Текст изложен на 190 страницах, содержит 16 рисунков и 50 таблиц. Список литературы содержит 133 источника, включая 11 отечественных и 122 — зарубежных.
СОДЕРЖАНИЕ РАБОТЫ
Материалы и методы исследования
Настоящая работа выполнена на кафедре медицинской генетики ГОУ ДПО «Санкт-Петербургской Медицинской Академии последипломного образования Федерального Агентства по Здравоохранению и социальному развитию», в МГЦ, в лаборатории Стабильности хромосом и клеточной инженерии Института цитологии РАН.
Обследовано 9 пациентов в возрасте от 2 до 17 лет с клинической картиной СВ с применением следующих методов: анамнестического, клинико-морфологического, цитогенетического и молекулярно-цитогенетического (флюоресцентная гибридизация на хромосомных препаратах (in situ), FISH), статистического, клинико-генеалогического. На основании анамнестического метода была собрана информация о физическом и психическом развитии больных детей, фенотипе «лицо эльфа», а также о ряде функциональных особенностей в каждом конкретном случае до проведения данного исследования. Клинико-морфологический метод включал антропометрию, детальное описание фенотипа по оригинальному плану (число типичных и дополнительных лицевых дисморфий, число признаков изменения соединительной ткани, опорно-двигательного аппарата, зрительного анализатора, особенности локомоции, некоторые показатели психо-эмоционального статуса), аускультацию, пальпацию, оценку степени выраженности вторичных половых признаков. Цитогенетическое исследование было проведено по общепринятой методике, которая включала следующие этапы: культивирование лимфоцитов периферической крови (72 часа), введение колхицина в стандартном разведении за 1,5 часа до окончания культивирования; обработка гипотоническим раствором (0,55% KCl) в течение 20 мин., фиксация смесью этилового спирта с ледяной уксусной кислотой (в соотношении 1:3), приготовление хромосомных препаратов, окрашивание по GTG-методу, микроскопирование. Молекулярно-цитогенетический метод был использован для исследования критического района 7q11.2 с помощью ДНК-зондов: 743 g06 (STS-маркер D7S489B), 893 b12 (STS-маркер D7S672), 880 а10 (STS-маркер D7S2549) а также для исследования прицентромерной области короткого плеча 7 хромосомы с применением ДНК-зонда 946 h08 (STS-маркер D7S494). Для проведения FISH использовались хромосомные препараты, полученные цитогенетическим методом с небольшой модификацией: высушивание производилось над пламенем спиртовки с минимальным тепловым воздействием; хранение в 70% этаноле в течение 10-14 дней при +4°С. В качестве ДНК-зондов был использован набор биотинилированных продуктов повторной амплификации фрагментов человеческого генома, первоначально находившихся в виде продукта первичной амплификации YAC (yeast artificial chromosome, искусственная дрожжевая хромосома). Процедура получения и очистки биотинилированных продуктов повторной амплификации была полностью проведена нами в лабораторных условиях.
Перечень и характеристики YAC, использованных для создания ДНК-зондов, представлены в таблице 1.
Для проведения молекулярно-цитогенетического исследования нам было предоставлено по одному образцу продукта первичной амплификации вставки человеческой ДНК в соответствующих шести YAC. Первичная амплификация (полимеразная цепная реакция, ПЦР) была проведена с помощью дегенеративного олигонуклеотидного праймера (ДОП) для всех использованных YAC и обозначалась как ДОП-ПЦР-1. На первом этапе исследования проводилась повторная амплифицикации с помощью ДОП (ДОП-ПЦР-2) для накопления материала по прилагавшемуся протоколу.
Таблица 1
Характеристики YAC, использованных для обследования пациентов с фенотипом синдрома Вильямса (опубликованы на интернет-сайте Max-Planck-Institute of Molecular Genetics, Берлин, Германия).
| Обозначение YAC | Локализация СГ* в геноме | Соответствующий STS-маркер | Локализация STS-маркера в 7 хромосоме, по данным: Sequence Map, п.н. / deCode Map, сМ / Radiation Hybrid Mapping | Длина, т.п.н. | Вторичные СГ* |
| 743 g06 | 7q11.2 | D7S489В | 71912480-71912902 — 394,7 cR3000 | 1640 | 5q14, 6q21 |
| 893 b12 | 7q11.2 | D7S672 | 70929546- 71307418 84,6-84,9 — | 720 | 11q12 |
| 880 a10 | 7q11.2 | D7S2549 | 62647936-64838974 77,91-79,6 — | 820 | — |
| 946 h08 | 7р11.1-7р11.2 | D7S494 | 57292183-57728878 76,71/78,49 239,8 cR3000 | 1280 | — |
| 660 g06 | 7p22 | D7S517 | 4271159-4561801 8,69 14,2 cR3000 | 130 | — |
| 965 c12 | 7q36 | D7S550 | 153556087-155017279 181 681,93 cR3000 | 160 | — |
* — сайт гибридизации
Продукты ДОП-ПЦР-2 были помечены с помощью биотин-16-2-дезоксиуридин-5-трифосфата по модифицированному протоколу для ДОП-ПЦР-2. Очистка меченых образцов осуществлялась методом фильтрации в колонках, заполненных сефадексом G-50, по стандартному протоколу. FISH проводилась по стандартному протоколу с модификациями времени денатурации ДНК хромосом и ДНК-зондов, а также предгибридизационной подготовки ДНК-зондов (эмпирический подбор количества Cot-1-ДНК, ДНК селезёнки крупного рогатого скота, обработанной ультразвуком). Для каждой реакции гибридизации использовался как минимум один ДНК-зонд для идентификации 7 хромосомы и всегда только один ДНК-зонд для исследования района, ответственного за СВ. Для иммунофлюоресцентной детекции биотина в красном свете использовались авидин, конъюгированный с Техасом красным, и биотинилированные козлиные моноклональные антитела к авидину (стандартный протокол). Окрашивание хромосом для визуализации производилось флюоресцентным красителем 4,6-диамидино-2-фенилиндол-2-HCl (стандартный протокол). Флюоресцентная микроскопия производилась с одновременным сохранением изображений в цифровом виде. Применяя метод FISH на контрольном материале (хромосомные препараты от 17 индивидуумов без клинических признаков СВ), мы провели оценку специфичности и чувствительности подготовленных нами ДНК-зондов, которые оказались адекватными задаче молекулярно-цитогенетического обследования больных с СВ. При обработке результатов FISH, проведённой на хромосомных препаратах, полученных от больных с СВ, мы применяли статистический анализ для установления достоверности полученных данных на предмет наличия либо отсутствия делеции сайта гибридизации для использованного ДНК-зонда у конкретного обследованного больного. Методом анализа было выбрано определение достоверности различия по критерию 2 между совокупностью метафазных пластинок в контрольном и опытном материале для каждого использованного ДНК-зонда. Поскольку задача анализа совокупности данных, полученных при исследовании больных с СВ и контрольной группы, является задачей анализа альтернативного распределения, анализируемые данные записывались в виде четырёхпольной таблицы в каждом случае применения данного ДНК-зонда у данного пациента. Число степеней свободы составило k=(S - 1)(r -1)=1, где S=2 — число граф (без итоговой графы), r=2 — число строк (без итоговой строки), 1 — константа. Поскольку во всех случаях как минимум в одной клетке четырёхпольной таблицы число наблюдений составляло величину, меньшую 4, для вычисления величины 2 была использована формула с поправкой Йетса:
2 = 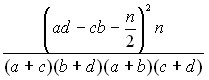 (1),
(1),
где а — число пластинок с одним сигналом исследуемого ДНК-зонда у больного с СВ; b — число пластинок с двумя сигналами ДНК-зонда у больного с СВ; c — число пластинок с одним сигналом ДНК-зонда в контрольной группе; d — число пластинок с двумя сигналами ДНК-зонда в контрольной группе; n = a + b + c + d.
Нулевая гипотеза была сформулирована следующим образом: число а появилось вследствие случайного стечения обстоятельств и, таким образом, отсутствует доказательство наличия делеции исследуемого STS-маркера (отсутствие сайта гибридизации (СГ) для данного ДНК-зонда, имеющего сайт гибридизации в сегменте 7q11.2 либо 7р11.1). Уровень значимости для вероятности -ошибки (р) был определён <0,05. Расчёт по формуле был выполнен с помощью компьютерной программы Maple 8.0. Поскольку во всех случаях k = 1, оценка полученного 2 проводилась по таблице граничных значений 2 для одной степени свободы с принятым уровнем р значимости <0,05. В тех случаях, когда полученное значение 2 превышало табличное значение 3,84, нулевая гипотеза была отвергнута, что подтверждало наличие делеции СГ данного ДНК-зонда (7q11.2/7р11.1) у данного больного. В тех случаях, когда полученное значение 2 было меньше табличного значения 3,84, нулевая гипотеза была подтверждена, а следовательно, было подтверждено отсутствие делеции исследуемого STS-маркера (наличие СГ для данного ДНК-зонда (7q11.2/7р11.1) в обоих гомологах 7 пары хромосом) у данного больного.
Применение формулы с поправкой Йетса (1) в ряде случаев привело к противоречивым результатам, когда полученное значение 2 превышало табличное значение 3,84 при заданном уровне р <0,05, в соответствии с чем нулевую гипотезу требовалось отвергнуть и, таким образом, подтвердить наличие делеции исследуемого STS-маркера. Однако в этих случаях формулируемый вывод противоречил фактическому результату исследования, при котором у обследованного не было зафиксировано ни одной пластинки с одним сигналом от данного ДНК-зонда, то есть возникала -ошибка (I рода). Отметим, что расчёт 2 по формуле для альтернативного распределения без поправки Йетса привёл к аналогичному противоречию во всех этих случаях. Для того чтобы не возникала -ошибка (I рода), нами в рассматриваемой группе случаях для статистической оценки данных был использован точный критерий Фишера, вычислявшийся по формуле:
![]() (2),
(2),
где а — число пластинок с одним сигналом исследуемого ДНК-зонда у больного с СВ; b — число пластинок с двумя сигналами от ДНК-зонда у больного с СВ; c — число пластинок с одним сигналом от ДНК-зонда в контрольной группе; d — число пластинок с двумя сигналами от ДНК-зонда в контрольной группе; n = a + b + +c + d. При получении значения р = р > 0,05 нулевая гипотеза не могла быть отвергнута.
Проведены сбор анамнеза (в том числе акушерско-гинекологический у матерей) и оценка фенотипа родителей больных детей по оригинальному плану (в том числе количественная оценка особенных черт лица).
Результаты исследования и их обсуждение
В настоящем исследовании у 9 больных с фенотипом, характерным для СВ, была изучена индивидуальная вариабельность типичных симптомов СВ, в частности такого фенотипического признака, как "лицо эльфа". Индивидуальная вариабельность последнего заключалась в различном количестве характерных дисморфий лица у пациентов разного возраста и пола: от 4 до 9 у конкретных больных.
В 8 из 9 случаев, когда проводилась эхокардиография, были диагностированы различные аномалии строения сердечно-сосудистой системы (ССС), среди которых наиболее распространённым оказался стеноз лёгочной артерии. Помимо типичной для СВ артериальной гипертензии (АГ), в исследованной группе была диагностирована стойкая артериальная гипотензия у больной женского пола.
У 58% больных нами отмечено изменение обмена кальция. У двух больных дошкольного возраста отмечена гиперкальцемия; у двух больных школьного возраста выявлена гипокальцемия, что может указывать на существование двух вариантов или двух фаз нарушения обмена кальция. Впервые выявлено сочетание гипокальцемии с артериальной гипертензией при СВ.
Нами отмечен ряд признаков моторной дисфункции желудочно-кишечного тракта (ЖКТ) у всех больных.
Впервые описано сочетание анамнестической поздней гиперкальцемии с хронической артериальной гипотензией и хроническим симптомокомплексом поражения ЖКТ (частые рвоты, запоры, резистентные к применявшейся симптоматической терапии, гипотония желчного пузыря и дискинезия желчевыводящих путей, сниженный аппетит).
Установлено, что на момент клинико-морфологического обследования все больные отставали в умственном развитии. Психо-моторное развитие в течение первых 2 лет жизни ни у одного из пациентов не соответствовало принятым в современной педиатрии нормам. У большинства больных отмечено нарушение локомотивных актов (89%) и недостаточное развитие мелкой моторики (78%).
Выявлено, что большинство больных отставало в физическом развитии, по данным анамнеза и/или клинико-морфологического обследования. Телосложение было оценено как типичное для СВ в половине случаев (45%), причём соотношение полов составило Ж:М=1:1. Установлено наличие не полностью характерного телосложения практически у половины больных (56%; с преобладанием мальчиков).
Степень поражения соединительной ткани варьировала у пациентов разного возраста и пола. Наибольшее количество проявлений её патологического строения составило 9 из 10 изученных признаков (у больного в возрасте 7 лет), наименьшее количество — 3 признака (4 пациента: в возрасте от 2 до 17 лет, Ж:М = 1:1). Сходные изменения зубов имели место у 2/3 больных. Мышечный тонус был оценен как сниженный у большинства больных (78%).
Все больные в исследованной группе имели изменения зрительного анализатора (ЗА). Кроме “звёздчатости” радужки и косоглазия, считающихся типичными для СВ, у отдельных больных выявлены снижение остроты зрения и нарушение цветового зрения, атрофический процесс в области диска зрительного нерва и мегалокорнеа.
Выявлено, что со стороны слухового анализатора у всех больных имелись те или иные специфические для СВ симптомы. У большинства больных были отмечены дизартрия (89%), эхолалия (89%), хорошая слуховая память (56%). Особенности речевого развития варьировали: от раннего его начала (первые слова в 7 мес.) с последующим отставанием вплоть до выраженной алалии в дошкольном возрасте.
У всех больных на момент проведения исследования отмечены такие психо-эмоциональные особенности, как привязанность и тёплое отношение к близким людям.
Нами отмечено некоторое опережение сверстников в половом развитии у больных детей, достигших препубертатного периода.
Таким образом, по данным настоящего исследования, клинические признаки СВ не ограничиваются типичными симптомами поражения ССС, ЦНС, ЗА, непостоянно встречающимися АГ и нарушениями обмена кальция и холестерина. К проявлениям СВ могут быть отнесены также:
- сниженный потенциал физического развития;
- патологическое строение соединительной ткани (которое является основой формирования целого ряда признаков — типичных лицевых дисморфий, голубоватой окраски склер, аномалий ССС, низкого голоса, типичного телосложения и изменений опорно-двигательного аппарата (ОДА), а также, возможно, характерных изменений зубов и снижения мышечного тонуса);
- гиперметропия как возможная частая аномалия рефракции;
- особенности локомотивных актов (нарушение передвижения по ступеням, боязнь резких движений), которые могут быть обусловлены дискоординацией движений в связи с поражением ЗА, ОДА, мозжечка, других отделов ЦНС;
- ряд особенностей речевого развития (раннее произнесение слов, позднее развитие фразовой речи, алалия, дизартрия);
- изменение функций ЖКТ;
- гипокальцемия.
В исследованной группе нами описан ряд микроаномалий организма, происхождение которых осталось неясным, в частности сочетание ВПР позвоночника с пилоростенозом в одном случае.
При цитогенетическом исследовании у одного больного был выявлен аномальный кариотип (перицентрическая инверсия 10 хромосомы). Остальные пациенты имели нормальный кариотип.
Перед проведением молекулярно-цитогенетического исследования СВ нами были исследованы специфичность и чувствительность ДНК-зондов, предназначенных для обследования больных с СВ путём проведения FISH на хромосомных препаратах, полученных от 17 индивидуумов, не имевших клинических признаков СВ. Согласно полученным результатам, каждый из использованных ДНК-зондов выявлял соответствующий его характеристике первичный СГ в 100% метафазных пластинок, имевших гибридизационный/ые флюоресцентный/ые сигнал(ы); их абсолютное число составило от 146 до 232 для конкретных ДНК-зондов. Чувствительность применявшихся ДНК-зондов составила от 65% до 91% для конкретных ДНК-зондов; абсолютное число исследованных метафазных пластинок составило от 312 до 686 для конкретных ДНК-зондов. Следует отметить, что при исследовании чувствительности ДНК-зондов нами не установлено наличия делеций соответствующих STS-маркеров у контрольных индивидуумов, поскольку в каждом конкретном случае доля метафазных пластинок с единственным ФС от исследуемого ДНК-зонда практически не превышала долю метафазных пластинок с единственным ФС от исследуемого ДНК-зонда в совокупности метафазных пластинок, полученных от соответствующей группы контрольных индивидуумов.
С помощью молекулярно-цитогенетического метода (FISH) и статистического анализа у 9 обследованных детей с типичной клинической картиной СВ нами было установлено с высокой степенью достоверности (p << 0,05) наличие делеции в пределах критического района 7q11.2, которая в 7 случаях представляла собой делецию STS-маркера D7S489B (отсутствие СГ для ДНК-зонда 743 g06). У 3 больных нами было установлено с высокой степенью достоверности (p << 0,05) наличие делеции STS-маркера D7S672 (отсутствие СГ для ДНК-зонда 893 b12), который, по данным литературы, в большинстве случаев остаётся интактным. В одном из этих случаев делеция STS-маркера D7S672 сочеталась с делецией STS-маркера D7S489B (отсутствие СГ для ДНК-зондов 743 g06 и 893 b12 одновременно). Делеции STS-маркера D7S2549 (ДНК-зонд 880 а10) выявить не удалось. Таким образом, были получены данные о разнородности молекулярного дефекта в пределах критического района для СВ.
Кроме того, молекулярно-цитогенетическое исследование прицентромерной области короткого плеча 7 хромосомы с высокой степенью достоверности (p << 0,05) показало наличие делеции STS-маркера D7S494 (отсутствие СГ для ДНК-зонда 946 h08) у 4 больных, во всех случаях — в сочетании с делецией STS-маркера D7S489B. Описание подобного феномена не встретилось нам при изучении литературы. Эти данные согласуются с гипотезой негомологичного кроссинговера как механизма возникновения делеций критического района при СВ (с учётом предположения о локализации проксимальной точки разрыва в коротком плече 7 хромосомы) и позволяют предположить существование феномена перицентрической инверсии 7 хромосомы при СВ, при которой разрывы локализуются в непосредственной близости от STS-маркеров D7S489B и D7S494.
Сопоставление клинических и молекулярно-цитогенетических данных выявило наличие поздней гиперкальцемии у 2 больных с делецией STS-маркера D7S494 (отсутствие СГ для ДНК-зонда 946 h08).
Установлено, что перинатальный период был неблагополучным у всех больных с СВ по 4-10 из 10 изученных признаков.
Изучение нами родословных больных с СВ не выявило нозологических единиц, которые можно было бы охарактеризовать как специфичные для генеалогического анамнеза при СВ.
ВЫВОДЫ
1. Типичные клинические проявления СВ характеризуются индивидуальной вариабельностью по числу лицевых дисморфий и признаков патологии костной и соединительной ткани, вариантам аномалий сердечно-сосудистой системы. В отдельных случаях характерный фенотип СВ может включать нетипичные врождённые пороки, микроаномалии и особенности, в том числе артериальную гипотензию, гипокальцемию.
2. Уровни специфичности и чувствительности использованных при молекулярно-цитогенетическом исследовании ДНК-зондов являются достаточными для выявления делеции соответствующих STS-маркеров (расположенных в пределах сайтов гибридизации конкретных ДНК-зондов). В контрольной группе не выявлено случаев делеции исследованных STS-маркеров.
3. Молекулярно-цитогенетический метод исследования (флюоресцентная гибридизация ДНК-зондов на хромосомных препаратах) является ценным и необходимым способом верификации клинического диагноза СВ. У всех больных в исследованной группе с высокой степенью достоверности (р<0,05) установлено наличие делеции в районе 7 хромосомы, ответственном за развитие клинической картины СВ. Протяжённость микроделеции в критическом районе 7 хромосомы имеет индивидуальную вариабельность.
4. Типичная делеция в критическом районе в некоторых случаях сочетается с делецией в области локализации STS-маркера D7S494 (отсутствие СГ для ДНК-зонда 946 h08) в коротком плече 7 хромосомы, за пределами района 7 хромосомы, ответственного за развитие клинической картины СВ.
5. Разработана система лабораторной диагностики СВ с применением оригинального набора ДНК-зондов, включающего ДНК-зонд 946 h08 с СГ в прицентромерной области короткого плеча 7 хромосомы (в области STS-маркера D7S494).
6. Фенотипических проявлений, строго специфичных для выявленных вариантов изменения структуры 7 хромосомы, установить не удалось.
Практические рекомендации.
Данные проведённого исследования свидетельствуют в пользу правомочности концепции множественного поражения организма при синдроме Вильямса и подтверждают представление об этом заболевании как о фенотипическом проявлении хромосомной микроделеции. Предлагаем следующий комплекс мероприятия при первичном обследовании больного ребёнка с клинической картиной синдрома Вильямса:
- кариотипирование;
- молекулярно-цитогенетическое исследование района 7 хромосомы, ответственного за СВ;
- эхографическое исследование сердца; применение рентгеноконтрастного метода — по показаниям; контроль артериального давления независимо от возраста (АД); консультация кардиолога/кардиохирурга;
- консультация невропатолога при выявлении патологических изменений, инструментальное обследование — по показаниям;
- консультация психоневролога (невропатолога) для выявления типичного интеллектуального “профиля” и особенностей координации движений;
- определение уровня кальция и параметров липидного обмена (холестерин, -липопротеиды) в крови независимо от возраста пациента;
- эхографическое исследование внутренних органов;
- консультация эндокринолога, обследование — по показаниям;
- консультация ортопеда;
- консультация офтальмолога с оценкой функции цветового зрения;
- консультация гастроэнтеролога;
- консультация генетика.
Предлагаем комплекс мероприятий при диспансерном наблюдении больных с синдромом Вильямса:
- контроль АД при плановых осмотрах педиатра, а также при наличии интеркуррентных заболеваний;
- при выявлении повышения АД — консультация кардиолога; по показаниям — мониторинг артериального давления, подбор корректирующей терапии;
- повторное эхографическое исследование сердца (по назначению кардиолога/кардиохирурга), внутренних органов (включая почки); при выявлении патологических изменений — консультация специалиста соответствующего профиля;
- наблюдение невропатолога;
- наблюдение психоневролога и разработка индивидуальных мероприятий по социальной реабилитации больного с учётом совокупности и степени выраженности характерных для СВ и индивидуальных особенностей пациента;
- наблюдение офтальмолога;
- при наличии патологии ОДА — наблюдение специалиста, коррекционные мероприятия — по показаниям;
- наблюдение гастроэнтеролога, подбор корректирующей терапии при моторной дисфункции ЖКТ.
Учитывая данные проведённого исследования, представляется целесообразным обследование членов ядерной семьи на этапе медико-генетического консультирования, что может послужить также предметом для дальнейшего исследования:
- кариотипирование и молекулярно-цитогенетическое обследование всех членов ядерной семьи;
- эхографическое исследование сердца и внутренних органов (включая почки) для всех членов ядерной семьи;
- оценка особенностей интеллекта у всех членов ядерной семьи специалистами соответствующего профиля;
- клинико-морфологический осмотр клиническим генетиком всех членов ядерной семьи;
- медико-генетическое консультирование семьи.
СПИСОК РАБОТ, ОПУБЛИКОВАННЫХ ПО ТЕМЕ ДИССЕРТАЦИИ
- А.Д. Прокофьева / Томилин Н.В., Василькова И.В., Прозорова М.В., Хитрикова Л.Е. Клиника и диагностика синдрома Вильямса у детей // Российский семейный врач. — 2002. — Т.6.— №2. — С. 13-19.
- A.D. Prokofyeva / I.V. Butomo, I.V. Vassilkova, T.A. Ledatshcheva, S.P. Maksimova, I.G. Pantova, M.V. Prozorova, L.E. Hitrikova, N.V. Tomilin. Clinical and molecular cytogenetical investigation of Williams syndrome in children. // 5th Balkanen Symposium Human Genetics. — Sofia, 2002. — P. 39-40.
- A.D. Prokofyeva. The results of the Williams syndrome clinical investigation in nine children confirm the multisystem disorder conception. // Материалы Международной научно-практической конференции "Врачи мира — пациентам". — СПб, 2003. — С. 45-46.
- А.Д. Прокофьева Результаты клинического исследования синдрома Вильямса у 9 детей города Санкт-Петербурга подтверждают концепцию множественного поражения организма при данном заболевании. // Медицинская генетика. Тезисы Всероссийской научно-практической конференции «Современные достижения клинической генетики». — Москва, 2003. — №10. — С. 438.
- А.Д. Прокофьева / Н.В. Томилин, М.В. Прозорова, И.В. Бутомо, И.В. Василькова, Т.А. Ледащева, С.П. Максимова, Г.И. Пантова, Л.Е. Хитрикова. Молекулярно-цитогенетическое исследование при синдроме Вильямса у детей из Санкт-Петербурга. // «Медицинская генетика». — 2003. — Т.2. — №11. — С. 469-473.
- А.Д. Прокофьева, Н.В. Томилин, Л.В. Соловьёва, М.В. Прозорова. Результаты молекулярно-цитогенетического обследования 9 детей с клинической картиной синдрома Вильямса: варианты делеций. // Сборник тезисов докладов первого съезда терапевтов Сибири и Дальнего Востока. — Новосибирск, 2005. — С. 594-596.