

Роль редокс-чувствительных мар-киназ jnk и р38 в дизрегуляции апоптоза мононуклеарных лейкоцитов при окислительном стрессе
На правах рукописи

Кайгородова Евгения Викторовна
РОЛЬ РЕДОКС-ЧУВСТВИТЕЛЬНЫХ МАР-КИНАЗ JNK И Р38 В ДИЗРЕГУЛЯЦИИ АПОПТОЗА МОНОНУКЛЕАРНЫХ ЛЕЙКОЦИТОВ ПРИ ОКИСЛИТЕЛЬНОМ СТРЕССЕ
14.00.16 – патологическая физиология
03.00.25 – гистология, цитология, клеточная биология
АВТОРЕФЕРАТ
диссертации на соискание ученой степени
кандидата медицинских наук
Томск – 2008
Работа выполнена в Государственном образовательном учреждении высшего профессионального образования «Сибирский государственный медицинский университет Федерального агентства по здравоохранению и социальному развитию»
Научные руководители:
доктор медицинских наук, профессор Рязанцева
Наталья Владимировна
доктор медицинских наук, профессор,
академик РАМН,
Заслуженный деятель науки РФ Новицкий
Вячеслав Викторович
Официальные оппоненты:
доктор медицинских наук, профессор Степовая
Елена Алексеевна
кандидат медицинских наук Солонский
Анатолий Владимирович
Ведущая организация: ГОУ ВПО Красноярская государственная медицинская академия Росздрава (г. Красноярск).
Защита состоится «___»____________2008 г. в _______часов на заседании диссертационного совета Д 208.096.01 при ГОУ ВПО Сибирский государственный медицинский университет Росздрава (634050, г. Томск, ул. Московский тракт, 2)
С диссертацией можно ознакомиться в научно-медицинской библиотеке ГОУ ВПО Сибирский государственный медицинский университет Росздрава
Автореферат разослан «___»___________2008 г.

Ученый секретарь
диссертационного совета Суханова Г.А.
ОБЩАЯ ХАРАКТЕРИСТИКА РАБОТЫ
Актуальность проблемы. Апоптоз представляет собой активную форму клеточной гибели, которая является физиологическим механизмом устранения избыточных и/или функционально неполноценных клеток. Нарушение реализации летальной программы клеток вследствие дисбаланса про- и антиапоптогенных факторов приводит к патологическим изменениям структуры и функций органов и тканей.
Важным звеном патогенеза различных заболеваний (онкологические, сердечно-сосудистые, нейродегенеративные, острые и хронические воспалительные процессы, сахарный диабет и др.) могут являться нарушения механизмов регуляции апоптоза, приводящие к его излишнему активированию или ингибированию [Bredesen D.E., 2000; Меньщикова Е.Б. и соавт., 2006; Жукова О.Б. и соавт., 2007]. В то же время развитие указанных заболеваний связано с повреждением клеток, обусловленным окислительным стрессом вследствие прооксидантно-антиоксидантного дисбаланса [Зенков Н.К. и соавт., 2001]. В роли повреждающих агентов выступают активные формы кислорода (АФК), которые являются эффективным инструментом локального действия за счет высокой реакционной способности. Нарастание содержания АФК приводит к окислительной модификации биомолекул, изменению активности ключевых ферментных систем, нарушению структуры мембран [Дубинина Е.Е., 2001; Меньщикова Е.Б. и соавт., 2006].
Наряду с этим избыточная генерация АФК в тканях на фоне истощения резервов антиоксидантной защиты оказывает влияние на функциональное состояние редокс-чувствительных систем внутриклеточной регуляции апоптоза. К числу последних относятся митоген-активируемые протеинкиназы JNK и р38, фосфорилирующие ответственные за реализацию летальной программы клеток белки-мишени [Gallo K.A., Johnson G.L., 2002].
Активация JNK играет ведущую роль в запуске летальной программы клеток в ответ на стресс (воздействие провоспалительных цитокинов IL-1, TNF- и свободных радикалов, образующихся под влиянием ультрафиолета и -радиации; ингибирование белкового синтеза) [Gallo K.A., Johnson G.L., 2002]. JNK индуцирует апоптоз путем фосфорилирования и активации фактора транскрипции Р53 [Моргункова А.А., 2005]. Установлено, что JNK-киназа может проникать в митохондрии, где фосфорилирует и активирует проапоптотические белки Bax и Bad, а также инактивирует антиапоптотические белки семейства Bcl-2 [Gallo K.A., Johnson G.L., 2002; Влаопулос С., Зумпурлис В.С., 2004; Дас Д.К., Молик Н., 2004; Teraishi F., Wu S., 2005; Harada C., Nakamura K., 2006]. Киназа р38 активирует фактор Nf-kB, MEF2C [Gallo K.A., Johnson G.L., 2002], способствует экспрессии и митохондриальной трансдукции одного из важнейших апоптогенных белков – Вах, опосредуя свое влияние через фосфорилирование Р53 [Kim S.J. et al., 2002; Mayr M. et al., 2002].
Вместе с тем, ряд исследований свидетельствуют о наличии антиапоптотической активности JNK и p38 [Sabapathy K. et al., 1999; Harada C., Nakamura K., 2006], зависящей от особенностей индуцирующих сигналов, комбинаций возможных путей их передачи и типов клеток. Расхождение представленных взглядов на обозначенную проблему затрудняет разработку фармацевтических подходов целенаправленной коррекции программированной гибели клеток и обусловливает целесообразность проведения исследования, направленного на изучение роли МАР-киназ в условиях окислительного стресса. В связи с этим возникает необходимость более подробного изучения роли стресс-активируемых протеинкиназ JNK и p38 в реализации летальной программы клеток при окислительном стрессе, являющемся типовым универсальным механизмом развития патологических процессов разного генеза.
Цель исследования: установить молекулярные механизмы регулирующего влияния редокс-чувствительных МАР-киназ на реализацию программированной гибели мононуклеарных лейкоцитов при окислительном стрессе.
Задачи исследования:
- Оценить уровень активных форм стресс-активируемых МАР-киназ (JNK и р38) в мононуклеарных лейкоцитах крови при дисбалансе окислительного метаболизма in vitro.
- Установить особенности реализации апоптоза мононуклеарных лейкоцитов крови при действии селективных ингибиторов JNK и р38 МАРК в условиях окислительного стресса in vitro.
- Определить роль редокс-чувствительных киназ JNK и р38 в модуляции фактора транскрипции Р53 при окислительном стрессе in vitro.
- Оценить участие редокс-чувствительных киназ JNK и р38 в продукции цитокинов (IL-8 и IL-10) мононуклеарными лейкоцитами крови в условиях окислительного стресса in vitro и при остром воспалении.
- Выявить молекулярные механизмы модуляции программированной гибели мононуклеарных лейкоцитов крови, опосредованные через действие редокс-чувствительных МАР-киназ, при остром воспалительном процессе.
Научная новизна. С помощью современных молекулярно-биологических методов исследования впервые проведена оценка роли редокс-чувствительных МАР-киназ p38 и JNK в нарушении реализации программированной гибели клеток в условиях окислительного стресса. Установлено, что в условиях дисбаланса окислительного метаболизма происходит активация редокс-чувствительных JNK и p38 МАР-киназ, являющихся важным элементом системы сигнальной трансдукции апоптогенных сигналов. При окислительном стрессе in vitro выявлена проапоптогенная роль регуляторных молекул JNK и р38. Установлено, что активация фактора транскрипции Р53 (за счет его фосфорилирования МАР-киназами и/или непосредственного действия АФК) приводит к изменению регуляции программированной гибели клеток в условиях окислительного стресса. В эксперименте с помощью селективного ингибитора SP600125 выявлено, что редокс-чувствительная киназа JNK влияет на продукцию IL-8, ингибитор р38 МАРК ML3403 не имеет такого эффекта. Впервые в эксперименте с использованием селективных ингибиторов МАР-киназ показано, что р38 и JNK не участвуют в регуляции синтеза IL-10 в условиях дисбаланса окслительного метаболизма.
Теоретическая и практическая значимость. Результаты проведенного исследования расширяют существующие представления о фундаментальных механизмах нарушения программированной гибели клеток при дисбалансе окислительного метаболизма. Получены приоритетные данные о роли редокс-чувствительных киназ JNK и р38 в дизрегуляции программированной гибели мононуклеарных лейкоцитов крови при экспериментальном окислительном стрессе и острых воспалительных заболеваниях, сопровождающихся усилением процессов свободно-радикального окисления. Полученные данные могут быть положены в основу разработки методологии коррекции нарушений регуляции апоптоза при патологических состояниях, сопровождающихся дисбалансом окислительного метаболизма.
Положения, выносимые на защиту:
- Дисбаланс окислительного метаболизма сопровождается активацией редокс-чувствительных систем сигнальной трансдукции апоптогенных сигналов, в частности активацией фосфорилирования МАР-киназ JNK и р38.
- В условиях окислительного стресса МАР-киназы JNK и р38 выполняют роль проапоптогенных регуляторных молекул.
- Активация фактора транскрипции Р53 в уловиях окислительного стресса in vitro обусловлена его фосфорилированием МАР-киназами и/или непосредственным эффектом АФК.
- При острых воспалительных заболеваниях (острый аппендицит, внебольничная пневмония) и экспериментальном окислительном стрессе in vitro нарушен баланс IL-8 и IL-10; повышение продукции IL-8 при нарушении окислительного метаболизма сопряжено с активацией JNK-киназы
Апробация и реализация работы. Результаты проведенных исследований докладывались и обсуждались на VIII Всероссийской научно-практической конференции «Актуальные вопросы клиники, диагностики и лечения, больных в многопрофильном лечебном учреждении» (Санкт-Петербург, 2007), III Всероссийской научно-практической конференции «Фундаментальные аспекты компенсаторно-приспособительных процессов» (Новосибирск, 2007), Межрегиональной научно-практической конференции «Актуальные проблемы медицины» (Абакан, 2007), VII конгрессе молодых ученых и специалистов «Науки о человеке» (Томск, 2007), Межгородской конференции молодых ученых «Актуальные проблемы патофизиологии» (Санкт-Петербург, 2007).
Исследования поддержаны Советом по грантам при Президенте РФ для поддержки ведущих научных школ РФ в рамках проекта «Молекулярные основы нарушения гомеостаза клеток при актуальных заболеваниях инфекционной и неинфекционной природы» (НШ-4153.2006.7), РФФИ - «Молекулярные механизмы управления программированной гибелью клеток с использованием регуляторных молекул» (№ 07-04-12150), а также выполнены в рамках ФЦНТП (проект «Разработка способов коррекции нарушений регуляции апоптоза клеток при патологических процессах в условиях окислительного стресса», государственный контракт № 02.442.11.7276 от 20.02.2006 г).
Основные результаты диссертационного исследования включены в лекционный курс по патологической физиологии («Патофизиология клетки», «Типовые патологические процессы», «Роль апоптоза клетки в патологии») для студентов лечебного и педиатрического факультетов ГОУ ВПО СибГМУ Росздрава.
Публикации. По материалам диссертации опубликовано 8 работ, из них 2 – в рецензируемых журналах, рекомендованном ВАК.
Структура и объем диссертации. Диссертация изложена на 152 страницах машинописного текста и состоит из введения, четырех глав, выводов и списка литературы, включающего 263 источника, из которых 105 - отечественных и 158 - иностранных. Работа иллюстрирована 6 таблицами и 20 рисунками.
ХАРАКТЕРИСТИКА ЭКСПЕРИМЕНТАЛЬНОГО И КЛИНИЧЕСКОГО МАТЕРИАЛА И МЕТОДЫ ИССЛЕДОВАНИЯ
Для выявления роли дисбаланса окислительного метаболизма в индукции апоптоза мононуклеарных лейкоцитов крови в качестве экспериментальной модели окислительного стресса in vitro в диссертационной работе было использовано инкубирование клеток здоровых доноров с различными концентрациями Н2О2. Кроме того, в работе были использваны мононуклеарные лейкоциты крови, полученные у больных острыми воспалительными заболеваниями (внебольничная пневмония и острый аппендицит), являвшихся примером патологического процесса, сопровождающегося дисбалансом окислительного метаболизма.
В диссертационной работе представлены результаты исследования мононуклеарных лейкоцитов крови, полученных у 34 здоровых доноров (18 мужчин и 16 женщин в возрасте от 18 до 55 лет) и у 49 больных (24 женщин и 25 мужчин в возрасте от 18 до 55 лет) с острыми воспалительными заболеваниями. Критериями включения пациентов в программу исследования являлись: отсутствие в анамнезе хронических инфекционных заболеваний, обострения хронических соматических заболеваний, а также злокачественных новообразований, психических расстройств, алкогольной и наркотической зависимости; возраст от 18 и до 50 лет; наличие информированного согласия.
Обследованные больные поступали в порядке скорой медицинской помощи в терапевтическое и хирургическое отделения ММЛПУ «Городская больница №1 (главный врач - С.М. Кирютенко), госпитальных клиник ГОУ ВПО СибГМУ Росздрава (главный врач – Заслуженный врач РФ, к.м.н. В.М. Шевелев), клиник Военно-Медицинского института (зав. кафедрой терапии и усовершенствования врачей – к.м.н., доц. Т.С.Агеева) г. Томска. Исследование проводилось при поступлении больных в стационар, что соответствовало периоду разгара болезни (1-3 сут от начала заболевания).
Материалом для исследования служила венозная кровь обследованных лиц, забор которой осуществляли утром натощак из локтевой вены в количестве 10 мл и стабилизировали гепарином (25 Ед/мл).
Исследование проводилось на базе Межкафедральной научно-образовательной лаборатории молекулярной медицины ГОУ ВПО СибГМУ Росздрава (зав. лабораторией – д.м.н. Л.С. Литвинова) и лаборатории клинической иммунологии ГУЗ ЦМСЧ № 81 ЗАТО г. Северск (зав. лабораторией – к.м.н. Т.Т. Радзивил).
В диссертационной работе были использованы методические подходы, направленные на оценку роли редокс-чувствительных киназ в регуляции апоптоза при окислительном стрессе. В таблице 1 представлено распределение здоровых доноров и больных острыми воспалительными заболеваниями в соответствии с использованными методами исследования.
Мононуклеарные лейкоциты выделяли из крови путем центрифугирования на слое фиколла (“Pharmacia” Швеция) плотностью 1,077. Выделенные мононуклеарные лейкоциты инкубировали в течение 18 ч при температуре 37С в полной питательной среде. Для определения роли митоген-активированных протеинкиназ р38 и JNK в регуляции апоптоза мононуклеарных лейкоцитов при окислительном стрессе использовали селективные ингибиторы МАР-киназ (ML3403 и SP600125 («Biosource», США) соответственно). Для индукции окислительного стресса в клеточные культуры добавляли перекись водорода в различных концентрациях (10 мкм, 50 мкм, 100 мкм, 500 мкм, 1 мМ и 5 мМ).
Уровень АФК и выраженность апоптоза в культуре мононуклеарных лейкоцитов определяли с помощью проточной лазерной цитометрии (Epics XL («Beckman Coulter», Франция)), для чего использовали дихлорфлюоресцеина диацетат (DCF-DA) («Sigma Aldrich», США) и FITC-меченный аннексин V («Catlag», США), соответственно. Содержание АФК в мононуклеарных клетках характеризовали в условных единицах (интенсивность свечения на клетку), активность апоптоза в культуре – в процентном содержании аннексин-положительных клеток. TUNEL-метод («Webstain», США) использовали для подтверждения наличия клеток с апоптотическими изменениями в исследуемых культурах мононуклеарных лейкоцитах. Содержание клеток со сниженным трансмембранным митохондриальным потенциалом определяли с использованием набора реагентов «MitoScreen» («BD Pharmigen», США) на проточном цитометре Epics XL («Beckman Coulter», Франция).
Определение содержания активных и неактивных форм МАР-киназ р38, JNK и фосфо-формы белка Р53 в мононуклеарных лейкоцитах крови проводили методом вестерн-блоттинга.
Клеточные экстракты получали путем лизиса клеток. Белки разделяли по молекулярной массе под действием электрического поля в течение 60 мин при напряжении 10 В/см пути. Для последующего исследования белки переносили на нитроцеллюлозную мембрану («Bio-Rad», США). Перенос белков осуществлялся электрофоретически в течение 90 мин при силе тока 60 мА. Мембраны последовательно инкубировали в TBST буфере с 5% обезжиренным сухим молоком и с первичными антителами к JNK 1 и 2, р38, к активным формам МАР-киназ (фосфо JNK 1 и 2, фосфо р38) и к фосфо-форме Р53 («Biosource», США) в разведении 1:200. Затем добавляли вторичные антитела с пероксидазной меткой (Biosource, США). В качестве стандарта и внутреннего контроля использовали белок глюкозо-3-фосфат-дегидрогеназу («Chemicon», США).
Таблица 1
Распределение здоровых доноров и больных острыми воспалительными заболеваниями в соответствии с использованными методами исследования
| Методы исследования | Группы обследованных (условия эксперимента) | ||||||
| Здоровые доноры | Культивирование клеток in vitro с 10, 50, 100, 500 мкМ, 1мМ и 5мМ Н2О2 | Культивирование клеток in vitro с 1мМ Н2О2 и селективными ингибиторами p38 ML3403 и JNK SP600125 | Культивирование клеток, полученных у больных с острыми воспалительными заболеваниями (внебольничная пневмония и острый аппендицит) | Культивирование клеток у больных с острыми воспалительными заболеваниями и селективными ингибиторами p38 ML3403 и JNK SP600125 | |||
| Проточная цитофлуориметрия | Содержание в клетке АФК | 34 | 82 | Не опреде-ляли | 49 | Не определяли | |
| Число апоптотически измененных клеток в аннексиновом тесте | 34 | 82 | 68 | 49 | 98 | ||
| Уровень митохондриального трансмембранного потенциала | 34 | 82 | Не опреде-ляли | 49 | Не определяли | ||
| Определение количества апоптотически измененных клеток TUNEL-методом | Не опреде-ляли | Не опреде-ляли | Не опреде-ляли | 10 | Не определяли | ||
| Определение содержание общих и фосфо-форм МАР-киназ (JNK, p38) и фосфо-Р53 методом вестерн-блоттинга | 4 | 4 | 4 | 4 | 4 | ||
| Исследование содержания IL-8 и IL-10 в супернатантах культур мононуклеарных лейкоцитов методом иммуноферментного анализа | 11 | 11 | 22 | 22 | 22 | ||
В супернатантах интактных, перекись-стимулированных и культивированных с ингибиторами МАР-киназ культур мононуклеарных лейкоцитов определяли содержание IL-8 и IL-10, используя метод твердофазного иммуноферментного анализа в соответствии с инструкцией к набору («Biosurce», USA) на микропланшетном фотометре Multiscan EX («ThermoLabSistems», Финляндия). Концентрации IL-8 и IL-10 вычисляли по калибровочной кривой.
Оценку полученных результатов проводили методами статистического описания и проверки статистических гипотез [Лакин А.В., 1989]. Проверку нормальности распределения количественных показателей проводили с использованием критерия Колмогорова-Смирнова. Для нормально распределенных выборок вычисляли средневыборочные характеристики: среднее арифметическое, среднее квадратичное отклонение, ошибка среднего. Для выборок, распределение которых отличалось от нормального, рассчитывали медиану, первый и третий квартили. При соответствии нормальному закону распределения признака в исследуемых выборках проверку гипотезы о равенстве средних выборочных величин проводили с использованием t-критерия Стьюдента. В случае отсутствия согласия данных с нормальным распределением для оценки различий между зависимыми выборками применяли непараметрический критерий Вилкоксона. Для оценки достоверности различий независимых выборок использовали ранговый критерий Манна-Уитни. При анализе использовали метод корреляционного анализа Спирмена. Различия считались достоверными при уровне значимости р<0,05 [Лакин А.В., 1989; Гланц С., 1999].
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ И ИХ ОБСУЖДЕНИЕ
Окислительный стресс является универсальным механизмом повреждения клетки при патологии разного генеза и характеризуется повышением внутриклеточной генерации активных форм кислорода (АФК) вследствие нарушения сбалансированности антиоксидантной и прооксидантной систем [Болдырев А.А., 2001]. Известно, что механизмы генерации АФК при многих патологических процессах носят типовой характер, хотя причины, вызывающие интенсификацию свободно-радикальных процессов, могут быть разными. При этом повышение внутриклеточной генерации активных форм кислорода приводит к окислительной модификации ряда регуляторных молекул.
Показано, что различные заболевания (онкологические, инфекционно-воспалительные, сердечно-сосудистые, нейродегенеративные и др.) сопровождаются дисбалансом окислительного метаболизма, с одной стороны, и нарушением программы апоптоза, – с другой [Меньщикова Е.Б. и соавт., 2006]. Последний необходим для устранения избыточных и/или функционально аномальных клеток, однако, нарушение реализации апоптоза играет важную роль в патологии человека. Так, угнетение клеточной гибели является одной из причин опухолевой трансформации и прогрессии, а ее излишняя стимуляция в нервной ткани ведет к развитию нейродегенеративных заболеваний [Барышников А.Ю., Шишкин Ю.В., 2002; Блохин Д.Ю., 2004; Моргункова А.А., 2005]. Дисбаланс про- и антиапоптогенных факторов, приводящий к изменению реализации программированной гибели, имеет место при воспалении, бактериальных и вирусных инфекциях [Пасечник А.В., 2004; Новицкий В.В. и соавт., 2006; Рязанцева Н.В. и соавт., 2007]. Выявление молекулярных путей нарушения реализации апоптоза в условиях окислительного стрессса – важнейшего механизма повреждения клетки при патологии разного генеза – было предметом нашего исследования. При этом мы сконцентрировали наше внимание на оценке роли редокс-чувствительных МАР-киназ в регуляции апоптоза мононуклеарных лейкоцитов при окислительном стрессе.
На первом этапе исследования для подтверждения положения о том, что дисбаланс окислительного метаболизма влияет на реализацию программированной клеточной смерти, мы применили общепринятый методологический подход моделирования окислительного стресса in vitro. В условиях эксперимента оценивали уровень внутриклеточной продукции АФК и количество апоптотически измененных клеток.
Одним из широко распространенных способов моделирования окислительного стресса in vitro является добавление в культуральную среду Н2О2 в различных конечных концентрациях [Abe J. et al., 1997; Griendling K.K. et al., 2000; Chen K., Vita J.A., 2001; Ding B., 2007].. Так, в культуре спленоцитов Т. Чанадири (2006) для индукции окислительного стресса использовал 100 мкМ Н2О2. В клетках эпидермальной карциномы человека А431 Е.Б. Бурова и соавт. (2003) инициировали окислительный стресс добавлением 1-2 мМ Н2О2. Известно, что в водных растворах в отсутствие каталазы, пероксидаз и ионов металлов переменной валентности Н2О2 относительно стабильна. Являясь электростатически нейтральной молекулой (не имея заряда), она легко проникает сквозь гидрофобные мембраны и может мигрировать в клетки и ткани [Choi Y.H., Furuse M., 1994]. Наличие нейтральных аддуктов Н2О2 (например гистидина) обеспечивает ее проникновение внутрь клеток даже в присутствии каталазы [Гамалей И.А.,1996].
В качестве модели окислительного стресса in vitro на культуре мононуклеарных лейкоцитов, полученных у здоровых доноров, мы использовали широкий диапазон концентраций Н2О2 - от 10 мкМ до 5 мМ. Исследование уровня активных форм кислорода показало, что в интактной культуре мононуклеарных лейкоцитов значения данного показателя составляли 0,24(0,19-0,33) усл.ед. на клетку. При добавлении в культуру клеток перекиси водорода в концентрации 10, 50, 100 и 500 мкМ уровень АФК в мононуклеарных лейкоцитах статистически не отличался от контроля, что могло быть связано с эффективной работой антиоксидантной системы. Достоверное увеличение содержания АФК в мононуклеарных лейкоцитах крови регистрировалось лишь при их культивировании с 1 мМ перекисью водорода (0,61(0,54-0,68) усл.ед), что свидетельствовало об изменении окислительного метаболизма в сторону истощения резерва антиоксидантной защиты (рис.1А) [Дубинина Е.Е.,2001; Меньщикова Е.Б. и соавт., 2006].
А) Б) 
Рис.1. Уровень активных форм кислорода (А), содержание апоптотических клеток и клеток со сниженным трансмембранным потенциалом митохондрий в общей популяции мононуклеарных лейкоцитов периферической крови (Б) в условиях культивирования in vitro с различными концентрациями перекиси водорода и у больных острыми воспалительными заболеваниями
Примечание: 1 - интактная культура мононуклеарных лейкоцитов; 2 - инкубирование клеток с 10 мкМ Н2О2; 3 - инкубирование клеток с 50 мкМ Н2О2; 4 - инкубирование клеток с 100 мкМ Н2О2; 5 - инкубирование клеток с 500 мкМ Н2О2; 6 - инкубирование клеток с 1 мМ Н2О2; 7 - мононуклеарные лейкоциты крови, полученные у больных внебольничной пневмонией; 8 - мононуклеарные лейкоциты крови, полученные у больных острым аппендицитом
Установлено, что большинство клеток устойчивы к воздействию экзогенной перекиси водорода в концентрации менее 50 мМ, однако добавление 0,1-50 мМ Н2О2 способно вызывать различные функциональные изменения в клетках, что позволяет рассматривать данный оксидант как важный внутриклеточный мессенджер [Гамалей И.А., Клюбин И.В., 1996; Тронов В.А., Константинов Е.М., 2000; Скулачев В.П., 2001]. В зависимости от концентрации АФК могут вызывать широкий спектр эффектов: регуляция клеточной пролиферации, индукция транскрипции определенных генов, цитотоксическое действие, запуск программы апоптоза [Haddad J.J., 2002; Шоф Н.Ф., Каган В.Е., 2004]. Насколько последний эффект реализуется в условиях культивирования мононуклеарных лейкоцитов с различными концентрациями перекиси водорода при возрастании уровня АФК в клетках, свидетельствуют данные, приведенные на рисунке 1Б.
Сравнительный анализ результатов аннексинового теста в культурах клеток, инкубированных с перекисью водорода в концентрациях 10, 50, 100 и 500 мкМ, не выявил достоверных изменений числа апоптозных клеток по сравнению с таковым в интактной культуре. Выраженная индукция летальной программы была обнаружена в культуре мононуклеарных клеток, подверженной воздействию 1 мМ перекиси водорода. При культивировании мононуклеарных лейкоцитов с 5 мМ Н2О2, помимо увеличения содержания апоптотических клеток, выявлялось резкое увеличение числа некротизированных мононуклеаров, что свидетельствовало о цитотоксическом эффекте Н2О2 в данной концентрации. Выявленные изменения при экспериментальном окислительном стрессе подтверждают факт участия АФК в индукции и передаче апоптотического сигнала.
Как было сказано выше, окислительный стресс является ключевым звеном патогенеза более чем ста заболеваний и патологических состояний (злокачественные новообразования, сердечно-сосудистые и нейродегенеративные заболевания, острые и хронические воспалительные процессы, сахарный диабет и др.) [Зенков Н.К., Ланкин В.З., 2001]. В клинической части нашего исследования влияния дисбаланса окислительного метаболизма на апоптоз клеток мы использовали мононуклеарные лейкоциты крови, полученные у пациентов с острыми воспалительными заболеваниями (острый аппендицит и внебольничная пневмония). Известно, что воспаление является одним из типовых патологических процессов, сопряженных с развитием дисбаланса окислительного метаболизма, выражающегося в увеличении продукции АФК клетками, усилении процессов перекисного окисления липидов и т.д. [Меньщикова Е.Б. и соавт., 2006]. Окислительный стресс играет важную роль в развитии всех трех фаз (альтерация, экссудация и пролиферация) острого воспаления. Среди АФК, продуцирующихся при окислительном стрессе, важную роль играет перекись водорода. Источниками Н2О2 служат ферментативные реакции с оксидазами, переносящими два электрона на молекулу кислорода (ксантиноксидаза, оксидаза L-аминокислот), а также реакция дисмутации, катализируемая СОД [Дубинина Е.Е., 2002]. Около 80% Н2О2, генерируемой фагоцитами в очаге воспаления, образуется в реакции дисмутации О2 супероксиддисмутазой [Панасенко О.М., Чеканов А.В., 2005]. Образованная при «дыхательном взрыве» перекись водорода может проникать в рядом расположенные клетки, вызывая в них увеличение продукции АФК за счет разобщения окислительного фосфорилирования. Распространяясь таким образом на значительные расстояния, в отсутствие прямых межклеточных контактов, Н2О2 приводит к изменениям структуры и функции клеток крови. Действительно, в проведенном нами исследовании отмечалось увеличение уровня АФК в мононуклеарных лейкоцитах крови у пациентов с внебольничной пневмонией и больных острым аппендицитом по сравнению с таковым в клетках у здоровых доноров (рис.1А).
В ходе настоящего исследования установлен факт нарушения реализации апоптотической гибели мононуклеарных лейкоцитов у пациентов с острыми воспалительными заболеваниями, аналогичного таковому при окислительном стрессе in vitro. У больных с острым аппендицитом и внебольничной пневмонией было выявлено усиление программированной смерти мононуклеарных лейкоцитов крови, соответствующее таковому в случае активационного апоптоза мононуклеарных лейкоцитов у здоровых доноров, вызванного добавлением в культуральную среду 1 мМ перекиси водорода (рис. 1Б). Влияние дисбаланса окислительного метаболизма на развитие апоптоза мононуклеарных лейкоцитов подтверждалось наличием положительной корреляционной связи между повышением уровня АФК и возрастанием количества апоптотически измененных мононуклеарных лейкоцитов при экспериментальном окислительном стрессе (r=0,84, p<0,05) и острых воспалительных заболеваниях (r=0,71, p<0,05). Увеличение активности процесса апоптоза в изученных культурах мононуклеаров, сопряженное с возрастанием уровня АФК в клетках, свидетельствует о вовлеченности редокс-чувствительных механизмов в регуляцию программированной гибели клеток.
Известно, что индукция программированной гибели клеток может быть связана с активацией как вне-, так и внутриклеточных путей ее запуска. Среди последних важную роль играет митохондриальный путь, так как в митохондриях сосредоточено большое количество проапоптотических факторов (цитохром с, Smac, AIF, эндонуклеаза G) [Joza N. et al., 2001; Cory S., Adams J.M., 2002]. В литературе встречается большое количество исследований, указывающих на взаимосвязь между генерацией АФК, функцией митохондрий и реализацией апоптоза [Брюне Б., 1998; Jackson M.J. et al., 2002; Андреев А.Ю. и соавт., 2005]. При этом митохондрии могут быть как мишенями регуляторных молекул в каскадах реакций, ведущих к апоптозу, так и генераторами АФК, используемых в данных каскадах в качестве сигнальных молекул [Green D.R., Reed J.C, 1998; Zhu H., Bunn H.F., 2001]. В проведенном нами исследовании снижение значения трансмембранного потенциала митохондрий было зафиксировано в мононуклеарных клетках, полученных у здоровых доноров и подвергнутых воздействию in vitro Н2О2 в концентрации 1 мМ, и у пациентов с внебольничной пневмонией и острым аппендицитом (рис. 1Б). Следует отметить, что при добавлении микромолярных концентраций перекиси водорода в культуру мононуклеарных лейкоцитов, полученных у здоровых доноров, наряду с отсутствием изменений митохондриального трансмембранного потенциала, число аннексин-положительных мононуклеаров также не изменялось (рис. 1Б).
Многие авторы считают, что уменьшение в результате повышения проницаемости наружной митохондриальной мембраны является критическим фактором для развития апоптоза [Kroemar G. et al., 1997; Regula K.M. et al., 2003]. В проведенном нами исследовании предположение об индукции апоптоза по митохондриальному пути в условиях окислительного стресса подтверждалось наличием положительной корреляционной связи между увеличением числа апоптотически измененных клеток и возрастанием количества мононуклеарных лейкоцитов со сниженным при инкубированнии in vitro с 1 мМ Н2О2 (r=0,78, p<0,05) и остром воспалении (r=0,69, p<0,05). Таким образом, можно предположить, что редокс-зависимое снижение является одним из элементов активации апоптогенных механизмов, отмеченной нами в условиях дисбаланса окислительного метаболизма.
Избыточная генерация активных форм кислорода в тканях на фоне истощения резервов антиоксидантной защиты оказывает влияние на функциональное состояние редокс-чувствительных систем внутриклеточной регуляции апоптоза. К числу последних относятся митоген-активируемые протеинкиназы JNK и р38, фосфорилирующие ответственные за реализацию летальной программы клеток белки-мишени, в том числе факторы транскрипции NF-kB, АР-1 и Р53 [Gallo K.A., Johnson G.L., 2002].
Активация JNK играет ведущую роль в запуске летальной программы клеток в ответ на стресс [Gallo K.A., Johnson G.L., 2002; Влаопулос С., Зумпурлис В.С., 2004]. Установлено, что JNK может индуцировать апоптоз путем фосфорилирования и активации фактора транскрипции Р53 [Моргункова А.А., 2005]. JNK-киназа проникает в митохондрии, где фосфорилирует и активирует проапоптотические белки Bax и Bad, а также инактивирует антиапоптотические белки семейства Bcl-2 [Gallo K.A., Johnson G.L., 2002; Влаопулос С., Зумпурлис В.С., 2004; Дас Д.К., Молик Н., 2004; Teraishi F., Wu S., 2005; Harada C., Nakamura K., 2006]. В апоптозе может участвовать субстрат JNK с-Мус [Baines C.P., Molkentin J. D., 2005; Cho S.D. et al., 2006]. Киназа р38 активирует фактор Nf-B и MEF2C [Gallo K.A., Johnson G.L., 2002], способствует экспрессии и митохондриальной трансдукции одного из важнейших апоптогенных белков – Вах, опосредуя свое влияние через фосфорилирование Р53 [Kim S.J. et al., 2002; Mayr M. et al., 2002]. Активируемые киназами транскрипционные факторы NF-kB и Р53, в свою очередь, контролируют синтез ключевых белков-регуляторов апоптоза. NF-kB стимулируют транскрипцию антиапоптотических генов Bcl-xL, X-IAP, c-IAP1 и c-IAP2, ингибируя тем самым летальную программу клеток [Wenger R.H., 2000].
Вместе с тем, ряд исследований свидетельствует о наличии антиапоптотической активности JNK и p38 [Sabapathy K. et al., 1999; Craig R. et al., 2000; Hoover H.E. et al., 2000; Andreka P. et al., 2001], зависящей от особенностей индуцирующих сигналов, комбинаций возможных путей их передачи и типов клеток. Показано, что трансфекция МКК6 (активирует р38 МАРК) усиливает антиапоптотическое действие через активацию NF-kB совместно с фосфорилированием -В-кристаллина, шаперона с известными защитными эффектами [Zechner D. et al., 1998; Craig R. et al., 2000; Hoover H.E. et al., 2000]. Согласно этим результатам, норэпинефрин-индуцированный апоптоз кардиомиоцитов возрастает при фармакологическом ингибировании МАР-киназы р38 [Communal C. et al., 2000].
Таким образом, JNK и р38-сигнальная трансдукция имеет как про-, так и антиапоптотические эффекты, хотя первые превалируют в большинстве экспериментальных моделей. В связи с этим возникает необходимость более подробного изучения роли стресс-активируемых протеинкиназ JNK и p38 в реализации летальной программы клеток при окислительном стрессе, являющемся универсальным механизмом развития патологических процессов разного генеза.
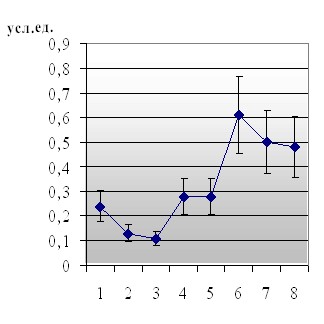
Раздел исследований, проведенных нами для выяснения роли JNK, р38 в регуляции программы апоптоза при окислительном стрессе, включал два этапа. На первом этапе использовался подход, основанный на оценке результатов эксперимента при избирательном блокировании функции киназ (в нашем случае исследование активности процесса апоптоза в культурах клеток, инкубируемых с селективными ингибиторами JNK и р38 (SP600125 и ML3403, соответственно)). Полученные данные свидетельствуют о том, что добавление ингибитора JNK (так же как и ингибитора р38) в культуру мононуклеарных лейкоцитов крови препятствовало увеличению числа аннексин-положительных клеток при окислительном стрессе in vitro и снижало их содержание у пациентов с острым воспалением (рис. 2).
Рис. 2. Содержание апоптотических клеток в общей популяции мононуклеарных лейкоцитов крови, при окислительном стрессе в условиях культивирования in vitro с ингибиторами МАР-киназ
Примечание: а - контроль; б - окислительный стресс in vitro; в - культивирование с 1 мМ Н2О2 и ингибитором ML3403; г - культивирование с 1 мМ Н2О2 и ингибитором SP600125; д - культивирование клеток, полученных у пациентов с острыми воспалительными заболеваниями; е - культивирование клеток, полученных у пациентов с острыми воспалительными заболеваниями, в условиях in vitro c ML3403; ж - культивирование клеток, полученных у пациентов с острыми воспалительными заболеваниями, в условиях in vitro c SP600125
Полученные в указанном аспекте данные позволяют считать, что в условиях дисбаланса окислительного метаболизма мононуклеарных лейкоцитов МАР-киназы JNK и р38 выступают в качестве проапоптогенных регуляторных молекул. Данное предположение согласуется с приводящимися в литературе сведениями о защитной роли ингибиторов р38 и JNK в случае сердечной дисфункции и апоптоза кардиомиоцитов, индуцированного ишемией [Meldrum D.R. et al., 1998; Ma X.L. et al., 1999; Barancik M. et al., 2000; Schneider S. et al., 2001]. Так, апоптоз кардиомиоцитов, индуцированный ишемией и доксорубицином в культуре, снижался при ингибировании р38 МАРК [Zhu W. et al., 1999; Kang Y.J. et al., 2000; Sharov V.G. et al., 2003]. V.L. Gabai et al. (2000) показали, что ингибирование JNK в Н9с2 миоцитах блокировало апоптоз, вызванный окислительным стрессом.
Следующим этапом исследования являлась попытка выявить молекулярные механизмы проапоптогенного эффекта МАР-киназ. При этом мы пытались получить ответы на ряд вопросов: сопряжена ли данная функция JNK и р38 с увеличением содержания в мононуклеарных лейкоцитах их активных (фосфорилированных) форм, которые могут оказывать воздействие на другие мишени - элементы сигнальной системы (факторы транскрипции, белки-регуляторы апоптоза); чем может быть обусловлено это увеличение – изменением общего содержания киназ при окислительном стрессе за счет увеличения их экспрессии, либо только активацией процесса фосфорилирования?
Результаты проведенной методом вестерн-блоттинга оценки содержания в мононуклеарных лейкоцитах общих и фосфорилированных форм JNK и р38 показала, что при окислительном стрессе, индуцированном добавлением 1мМ Н2О2 в культуру клеток, полученных у здоровых доноров, общее содержание JNK (подклассы JNK1 и JNK2) не изменялось по сравнению с контролем (рис. 3). Аналогичные результаты были получены и в клинике острого воспаления – общий уровень JNK и р38 в мононуклеарных лейкоцитах в этом случае соответствовал норме. Вместе с тем содержание фосфорилированных форм JNK и р38 увеличивалось (по отношению к контролю) при инкубации мононуклеарных лейкоцитов, полученных у здоровых доноров, с 1мМ Н2О2, и в клетках крови у пациентов с острыми воспалительными заболеваниями (рис. 3). Полученные данные свидетельствуют о том, что при окислительном стрессе увеличение уровня фосфорилированных форм редокс-чувствительных киназ JNK и р38 обусловлено их активацией АФК и не связано с изменением активности экспрессии данных ферментов в клетке.
АФК могут влиять на активность JNK и р38 посредством различных реакций [Brumell J.H., 1996; Турпанов К.Т., 2002; Меньщикова Е.Б. и соавт., 2006]. АФК активируют белки MAPK Kinase Kinase (в частности, белок ASK1- Apoptosis signal-regulating kinase 1, активирующий как JNK, так и р38), которые запускают сигнальный каскад [Tobiume K. et al., 2001; Matsuzawa A., Ichijo H., 2005]. JNK удерживается в неактивной форме глутатион-S-трансферазой класса Pi (GSTPi). Под действием Н2О2 происходит диссоциация этого комплекса и активация киназы JNK [Mathers J. et al., 2004; Меньщикова Е.Б. и соавт., 2006]. Гидроксиноненаль - конечный продукт перекисного окисления липидов - образует аддукт с JNK, вызывая ее активацию [Дубинина Е.Е., 2001]. АФК инактивируют фосфатазы ферменты, отщепляющие фосфатные группы от специфических ферментов, вызывая тем самым их инактивацию [Klein J.A, Ackerman S.L., 2003; Влаопулос С., Зумпурлис В.С., 2004]. Повышенный уровень АФК часто коррелирует с активацией фосфорилирования JNK и р38 [Baines C.P. et al., 2005; Gautam D.K. et al., 2005; Teraishi F. et al., 2005; Cho S.D. et al., 2006], что подтверждается результатами настоящего исследования (рис.1А, 3).
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | |
 p38 p38 | ||||||||||||
| P-p38 | ||||||||||||
| JNK2 JNK1 | ||||||||||||
| P-JNK2 P-JNK1 | ||||||||||||
| G3PDH |
Рис. 3. Уровень активных и неактивных форм МАР-киназ JNK и р38, определенный методом вестерн-блоттинга, в культурах мононуклеарных лейкоцитов, полученных у здоровых доноров (1-4 – интактная культура клеток, 5-8 – культура клеток после воздействия 1 мМ Н2О2) и у пациентов с внебольничной пневмонией (9-12) (G3PDF-глюкозо–3–фосфатдегидрогеназа)
Важнейшими сигнальными молекулами, активирующимися МАР-киназами и АФК непосредственно и участвующими в реализации летальной программы клеток, являются транскрипционные факторы. К числу последних относится Р53, также действующий как эффекторный проапоптогенный белок. В связи с этим особый интерес, на наш взгляд, представляет оценка содержания фосфо-формы Р53 и его влияния на регуляцию апоптоза мононуклеарных лейкоцитов при окислительном стрессе.
 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| P-P53 |
Рис.4. Уровень фосфо-формы Р53, определенный методом вестерн- блоттинга, в культурах мононуклеарных лейкоцитов в условиях окислительного стресса при ингибировании МАР-киназ р38 и JNK in vitro
Примечание: 1-4 – клетки, инкубированные с 1мМ Н2О2; 5-8 – клетки, инкубированные с 1мМ Н2О2 и ингибитором ML3403; 9-12 – клетки, инкубированные с 1мМ Н2О2 и ингибитором SP60012
С помощью вестерн блоттинга нами было выявлено наличие фосфо-формы Р53 в мононуклеарных лейкоцитах крови при экспериментальном окислительном стрессе in vitro (рис. 4).
Р53 активируется в ответ на различные стрессовые сигналы (УФ- радиация, гипоксия, окислительный стресс, дефицит глюкозы), которые приводят к повышению его концентрации в цитоплазме [Kastan M.B. et al., 1991; Gudkov A.V., Komarova E.A., 2003; Oren M., 2003].
В активации белка Р53 участвуют редокс-чувствительные киназы JNK и р38. JNK фосфорилирует опухолевый супрессор Р53 по Thr8. Когда JNK неактивна, она связывается с Р53 на участке между остатками 97 и 116, что приводит к деградации белка в протеосомах [Влаопулос С., Зумпурлис В.С., 2004]. Показано, что активация р38 через прямое фосфорилирование увеличивает содержание белка Р53, который вторично усиливает апоптоз, индуцируя экспрессию и митохондриальную трансдукцию Вах [Kim S.J. et al., 2002; Mayr M. et al., 2002].
Для выявления молекулярных механизмов участия JNK и р38 МАР-киназы в регуляции программированной клеточной смерти при окислительном стрессе мы определяли уровень фосфо-формы белка Р53 в культурах мононуклеаров у здоровых доноров, инкубированных с 1 мМ Н2О2 и ингибиторами МАР-киназ SP600125 и ML3403. В результате настоящего исследования было показано, что при культивировании мононуклеарных лейкоцитов с ингибиторами JNK и р38 в условиях окислительного стресса in vitro имеет место снижение содержания фосфо-формы белка Р53. Следует заметить, что применение как ингибитора JNK (SP600125), так и р38 (ML3403) предотвращало запуск апоптогенной программы, индуцированной окислительным стрессом. Полученные результаты подтверждают данные литературы об участии редокс-чувствительных киназ JNK и р38 в Р53-опосредованном апоптозе.
Поскольку АФК-индуцируемая активность JNK и р38 МАРК часто играет существенную роль в судьбе клетки, нас также заинтересовало участие данных киназ в продукции цитокинов мононуклеарными лейкоцитами в условиях окислительного стресса и при остром воспалении.
Одним из широко распространенных провоспалительных цитокинов является интерлейкин-8. IL-8 продуцируется многими типами клеток (моноцитами, нейтрофилами, эндотелиальными клетками, митоген-стимулированными Т-лимфоцитами) и обладает выраженными провоспалительными свойствами. Основным биологическим эффектом IL-8 является индукция хемотаксиса нейтрофилов, эозинофилов, базофилов, моноцитов и других клеток системы иммунитета. IL-8 усиливает ангиогенез in vivo и in vitro. Повышенный уровень IL-8 ассоциируется с хроническими и острыми воспалительными состояниями [Hack C., 1992]. Данный интерлейкин продуцируется под воздействием бактериальных эндотоксинов и цитокинов, главным образом, TNF и IL-1. Окислительный стресс может индуцировать также продукцию IL-8, активируя ядерные факторы транскрипции. Показано, что редокс-чувствительные транскрипционные факторы, такие как NF-kB и АР-1 активируются в клетках, участвующих в воспалении, приводя к ап-регуляции некоторых провоспалительных генов [Lakshminarayanan V. et al., 1998; Aydin M. et al., 2007].
В результате проведенного нами иммуноферментного анализа содержания провоспалительного цитокина IL-8 в супернатантах исследованных культур мононуклеаров было показано, что уровень IL-8 превышал контрольные значения в случае экспериментальной модели окислительного стресса in vitro и при остром воспалении. Кроме того, было установлено, что редокс-чувствительная киназа JNK влияет на продукцию IL-8, что выявлено в эксперименте с использованием селективного ингибитора SP600125. Ингибитор р38 МАРК ML3403 не имел такого эффекта (рис. 5).

Рис. 5. Содержание IL-8 в супернатантах культур мононуклеарных лейкоцитов при ингибировании МАР-киназ р38 и JNK в условиях окислительного стресса in vitro и при остром воспалении
Примечание: а - контроль; б - окислительный стресс in vitro; в - культивирование клеток с 1 мМ Н2О2 и ингибитором JNK SP600125; г – культивирование клеток с 1 мМ Н2О2 и ингибитором р38 ML3403; д - интактная культура клеток у больных острым аппендицитом; е - интактная культура клеток у больных внебольничной пневмонией; ж - инкубирование клеток у больных острыми воспалительными заболеваниями с ингибитором JNK SP600125; з - инкубирование клеток у больных острыми воспалительными заболеваниями с ингибитором р38 ML3403
По данным литературы, ингибитор JNK SP600125 блокирует экспрессию мРНК IL-8 и уменьшает продукцию данного цитокина различными клетками (эндотелиоциты, альвеолоциты А549, бронхиальные эпителиоциты человека) [Li L.F. et al., 2003; Saatian B. et al., 2006]. Высокая концентрация АФК в месте воспаления индуцирует МАР-киназу JNK, ускоряющую активацию воспалительных медиаторов и уничтожает посредством апоптоза клетки, утратившие способность к регуляции клеточного цикла. Для экспрессии воспалительных медиаторов — цитокинов, металлопротеиназ и адгезивных молекул - необходима активация JNK в присутствии АФК [Kathleen A., Johnson G. L., 2002; Влаопулос С., Зумпурлис В.С., 2004].
Регуляция IL-8 МАР-киназами отличается в зависимости от природы стимулов [Shapiro L., Dinarello C.A., 1995; Li L.F. et al., 2003; Kim M.S. et al., 2005; Harimaya A. et al., 2007] и типа клетки [Hashimoto S. et al., 1999; Li J. et al., 2002; Oudin S., Pugin J., 2002; Li L.F. et al., 2003; Aydin M. et al., 2007]. Окислительный стресс, вызванный экзогенной Н2О2, индуцирует синтез IL-8 в эпителиальных и эндотелиальных клетках [Lakshminarayanan V. et al., 1997; Shimada T. et al., 1999]. Этот факт подтверждают и результаты проведенного нами эксперимента. После культивирования мононуклеарных лейкоцитов крови, полученной у здоровых доноров, с 1 мМ Н2О2 определялось, как было показано выше, повышенное содержание IL-8 в супернатантах клеток (рис. 5).
Роль р38 МАР-киназы в регуляции синтеза IL-8 неоднозначна. Так, по данным ряда авторов, ингибитор р38 МАРК (SB 203580) значительно снижает продукцию данного цитокина, опосредуя свое действие через NF-kB [Kim M.S. et al., 2005; Saatian B. et al., 2006; Harimaya A. et al., 2007]. Напротив, L.F. Li, et al. (2003) показали, что ингибирование р38 МАРК не вызывает снижение экспрессии и секреции IL-8, предположив, что регуляция данных процессов осуществляется через активацию АР-1, NF-kB, зависящую от JNK и NIK (NF-kB-inducing kinase).
Таким образом, роль стресс-активированных киназ JNK и р38 в экспрессии и секреции клетками IL-8 в условиях дисбаланса окислительного метаболизма весьма неоднозначна и требует дальнейшего исследования.
Еще одним важным цитокином, продуцируемым мононуклеарными лейкоцитами крови при острых воспалительных заболеваниях, сопровождающихся окислительным стрессом, является интерлейкин-10.
IL-10 (фактор, ингибирующий синтез цитокинов) принадлежит к группе противовоспалительных медиаторов, блокирующих эффекты лимфоцитарных и макрофагальных провоспалительных цитокинов, а также подавляющих функцию антигенпрезентирующих клеток. IL-10 синтезируется Тh-2 типа, В-лимфоцитами, моноцитами и эпителиальными клетками [de Vries J.E., 1995; Foey A.D. et al., 1998; Игонин А.А. и соавт., 2004; Рубцова И.Е. и соавт., 2004]. Установлено, что IL-10 стимулирует экспрессию в моноцитах растворимых рецепторов TNF (sTNF) (естественных ингибиторов TNF) [Joyce D. A. et al., 1994] и увеличивает продукцию IL-1R антагониста (IL-1Rа), который конкурентно ингибирует связывание IL-1 с мембранным рецептором [de Waal Malefyt R. et al., 1993; Jenkins J.K. et al., 1994]. По данным литературы, IL-10 не оказывает прямого ростового действия на лимфоциты. Что же касается влияния IL-10 на апоптоз, то известно, что IL-10 способен его подавлять [Go N.F. et al., 1990]. Имеются многочисленные сведения о способности этого цитокина ингибировать апоптоз В-лимфоцитов, активированных Т-клеток [Pawelec G. et al., 1996; Cohen S.B. et al., 1997; Рубцова И.Е. и соавт., 2004].
В результате проведенного исследования нами было выявлено, что уровень IL-10 в супернатантах культур мононуклеарных лейкоцитов периферической крови, полученной у больных с острыми воспалительными заболеваниями, не отличался от контрольных значений. Аналогичные изменения данного показателя были зарегистрированы и при культивировании мононуклеарных лейкоцитов с 1 мМ Н2О2. С точки зрения изменения регуляции программы апоптоза мононуклеров, выявленное нами на первом этапе настоящего исследования увеличение количества аннексин-положительных клеток можно также объяснить отсутствием изменения синтеза IL-10, способного подавлять апоптоз [Cohen S.B. et al., 1997].
Для выяснения роли р38 и JNK МАР-киназ в регуляции синтеза IL-10 в условиях окислительного стресса мы использовали их селективные ингибиторы (ML3403 и SP600125, соответственно). В результате настоящего исследования было показано, что в условиях дисбаланса окислительного метаболизма ни р38, ни JNK не влияют на продукцию IL-10 мононуклеарами. Отсутствие изменений синтеза IL-10 в условиях окислительного стресса и при ингибировании МАР-киназ наводит на мысль об отсутствии участия обозначенных редокс-сигнальных систем в продукции данного цитокина.
Таким образом, результаты проведенного нами исследования расширяют существующие фундаментальные представления о характере изменения программы гибели клеток при дисбалансе окислительного метаболизма. Получены новые данные о роли МАР-киназ JNK и р38 в дизрегуляции программированной гибели мононуклеарных лейкоцитов крови при заболеваниях, сопровождающихся усилением процессов свободно-радикального окисления (внебольничная пневмония и острый аппендицит), а также в условиях окислительного стресса in vitro. Представленные нами результаты позволяют говорить о редокс-чувствительных киназах р38 и JNK как о возможных терапевтических мишенях коррекции нарушений апоптотической программы (в случаях её гиперактивации) в условиях окислительного стресса.
Выводы
- Инициация программированной гибели мононуклеарных лейкоцитов в условиях возрастания содержания в клетках активных форм кислорода сопряжена с индукцией митохондриального пути её запуска и увеличением уровня фосфо-форм МАР-киназ JNK и р38.
- Окислительный стресс индуцирует активацию системы сигнальной трансдукции, осуществляемой редокс-чувствительными МАР-киназами JNK и р38.
- Добавление селективных ингибиторов JNK и р38 (SP600125 и ML3403, соответственно) в культуру мононуклеарных лейкоцитов периферической крови у здоровых доноров препятствует увеличению числа аннексин-положительных клеток при окислительном стрессе in vitro.
- В условиях дисбаланса окислительного метаболизма мононуклеарных лейкоцитов МАР-киназы JNK и р38 выступают в качестве проапоптогенных регуляторных молекул.
- Активация фактора транскрипции Р53 в условиях дисбаланса окислительного метаболизма в клетках обусловлена его фосфорилированием МАР-киназами и/или непосредственным эффектом АФК.
- Ингибирование редокс-чувствительной киназы JNK приводит к снижению продукции IL-8 мононуклеарными лейкоцитами в условиях окислительного стресса in vitro. МАР-киназы р38 и JNK не участвуют в регуляции синтеза IL-10 в условиях дисбаланса окислительного метаболизма.
- В экспериментальной модели окислительного сресса in vitro и у больных с острыми воспалительными заболеваниями (острый аппендицит, внебольничная пневмония), сопровождающимися дизрегуляцией окислительного метаболизма, молекулярные механизмы реализации апоптотической программы мононуклеарных лейкоцитов крови, опосредованные редокс-зависимыми МАР-киназами, являются однотипными.
СПИСОК РАБОТ, ОПУБЛИКОВАННЫХ ПО ТЕМЕ ДИССЕРТАЦИИ
- Стресс-активируемая протеинкиназа JNK-молекулярная мишень для коррекции дизрегуляции апоптоза при патологических состояниях, сопровождающихся дисбалансом окислительного метаболизма / Н.Ю. Часовских, Е.В. Кайгородова, Е.Г. Старикова и др. // Материалы VIII Всероссийской научно-практической конференции «Актуальные вопросы клиники, диагностики и лечения, больных в многопрофильном лечебном учреждении» - г. Санкт-Петербург, 24-25 апреля 2007.- Вестник Российской военно-медицинской академии (приложение). – Санкт-Петербург. – 2007. – С. 497.
- Ингибиторы митоген-активированных протеинкиназ p38 и JNK как молекулярный механизм коррекции нарушений программированной гибели клеток при воспалении / Н.Ю. Часовских, Е.В. Кайгородова, Е.Г. Старикова и др.// Материалы VIII Всероссийской научно-практической конференции «Актуальные вопросы клиники, диагностики и лечения, больных в многопрофильном лечебном учреждении» – г. Санкт-Петербург, 24-25 апреля 2007.- Вестник российской военно-медицинской академии (приложение). – Санкт-Петербург. – 2007. – С. 497-498.
- Часовских, Н.Ю. Влияние окислительного стресса на реализацию апопототической программы мононуклеаров периферической крови в условиях in vitro / Н.Ю. Часовских, Е.Г. Старикова, Е.В. Кайгородова // Материалы межгородской конференции молодых ученых «Актуальные проблемы патофизиологии» - г. Санкт-Петербург, 24-25 апреля 2007. – Санкт-Петербург, 2007. – С. 139.
- Часовских, Н.Ю. Роль протеинкиназ Р38 и JNK (С-Jun NH2-terminal kinases) в дизрегуляции апоптоза мононуклеарных лейкоцитов в условиях окислительного стресса in vitro / Н.Ю. Часовских, Е.В. Кайгородова, Е.Г. Старикова / Материалы межгородской конференции молодых ученых «Актуальные проблемы патофизиологии» - г. Санкт-Петербург, 24-25 апреля 2007. - Санкт-Петербург – 2007. – С. 140.
- Кайгородова, Е.В. Роль митоген-активируемых протеинкиназ JNK(С-Jun NH2-terminal kinases) и р38 в регуляции апоптоза мононуклеаров периферической крови в условиях окислительного стресса in vitro и при остром воспалении / Е.В. Кайгородова, Н.Ю. Часовских // Материалы VII конгресса молодых ученых и специалистов «Науки о человеке» Томск, 17-18 мая 2007. – Томск. – 2007. – С.182-183.
- Роль протеинкиназ JNK и р38 в реализации программы апоптоза мононуклеаров в условиях окислительного стресса in vitro / Н. Ю.Часовских, Е.В. Кайгородова, Е.Г. Старикова, Ю.В. Стариков, Н.В. Рязанцева, В.В. Новицкий // Материалы III всероссийской научно-практической конференции «Фундаментальные аспекты компенсаторно-приспособительных процессов» - Новосибирск, 7 – 9 ноября 2007. – Медико-фармацевтический журнал. – 2007. – С.84 - 85
- Модуляция апоптоза мононуклеаров в условиях окислительного стресса / В.В. Новицкий, Н.В. Рязанцева, Н.Ю. Часовских, Е.В. Кайгородова и др. // Бюллетень экспериментальной биологии и медицины. – 2008. – №3. – С. 251-254.
- Роль митогенактивированных протеинкиназ JNK и р38 в регуляции апоптоза мононуклеаров крови в условиях окислительного стресса in vitro / Н.В. Рязанцева, В.В. Новицкий, Н.Ю. Часовских, Е.В. Кайгородова и др. // Бюллетень экспериментальной биологии и медицины. – 2008. – №5. – С. 505-509.
СПИСОК СОКРАЩЕНИЙ
АФК – активные формы кислорода
FITC - флюоресцеинизотиоцианат
ASK1 – apoptosis signal-regulating kinase 1
IL – интерлейкин
JNK – c-Jun N-terminal kinase
МАРК – митоген-активируемые протеинкиназы
Автор выражает благодарность зав. кафедрой терапии и усовершенствования врачей Военно-медицинского института к.м.н. Т.С. Агеевой, зав. терапевтическим отделением ММЛПУ «Городская больница №1» к.м.н. А.В Дубоделовой, зав. хирургическим отделением ММЛПУ «Городская больница №1» А.Я. Митасову, д.м.н., профессору кафедры госпитальной хирургии ГОУ ВПО СибГМУ Е.Г. Соколовичу за помощь в наборе клинического материала, ассистенту кафедры Фундаментальных основ клинической медицины ГОУ ВПО СибГМУ Росздрава к.м.н Н.Ю. Часовских и зав. лабораторией клинической иммунологии ГУЗ ЦМСЧ № 81 ЗАТО Северск к.м.н Т.Т. Радзивил за ценные теоретические советы и помощь в организации проведения исследований.